AGRICULTURAL MATERIALS
Development of Optimized
Extraction Methodology for Cyanogenic Glycosides from Flaxseed (Linum usitatissimum)
VÉRONIQUE J. BARTHET and RAY BACALA
Canadian Grain Commission, Grain Research Laboratory, 1404-303 Main St,
Winnipeg, MB, Canada, R3C 3G8
A reference method (higher accuracy) and a
routine method (higher throughput) were developed for the extraction of
cyanogenic glycosides from flaxseed. Conditions of (essentially) complete
extraction were identified by comparing grinding methods and extraction solvent
composition, and optimizing solvent-to-meal ratio, extraction time, and repeat
extraction. The reference extraction method consists of sample grinding using a
high-speed impact plus sieving mill at 18 000 rpm with a
1.0 mm
sieve coupled with triple-pooled extraction in a sonicating water bath (40°C, 30 min)
using 75% methanol. The routine method differs by the use of a coffee mill to
grind samples and a single extraction. The 70 and 80% methanol solutions were
equal and superior to other combinations from 50 to 100% aqueous ethanol or
methanol. The extraction efficiencies of the routine method (relative to the
reference method) was 87.9 ± 2.0% SD (linustatin) and 87.6 ± 1.9% SD
(neolinustatin) using four composite samples that were generated from seeds of
multiple cultivars over two crop years and locations across Western Canada.
Ground flaxseed was stable after storage at room temperature, refrigeration, or
freezing for up to
7 days, and frozen for at least 2 weeks but less than 2 months. Extracts
were stable for up to
1 week at room temperature and at least 2 weeks
when refrigerated or frozen.
laxseed (Linum
usitatissimum L.) contains the Fcyanogenic glycosides linustatin (2-[6-b-D-glucosyl-b-
D-glucopyranosyloxy]-2-methylpropionitrile)
and neolinustatin ((R)-2-[6-b-D-glucosyl-b-D-gluco-
pyranosyloxy] -2-methylbutyronitrile) in significant amounts (1). The
corresponding monoglycosides linamarin (2-b-D-glucopyrano-syloxy-2-methylpropionitrile) and lotaustralin ((R)-2-b-D-glucopyranosyloxy-2-methylbutyronitrile) are present in immature seed
(2, 3), but diminish to trace levels in mature seed (3, 4). Cyanogenic
glycosides are retained in meal after
Received March 27, 2009. Accepted by EB August 14, 2009. Corresponding
author’s e-mail: veronique.barthet@grainscanada.gc.ca
oil extraction and readily liberate cyanide upon
acid hydrolysis and by endogenous seed enzymes. As a result, the usefulness of
flaxseed and flaxseed meal in livestock and poultry feed products is limited.
Extraction and analytical methods for cyanogenic
glycosides from flax have been recently reviewed (5). Analytical assays fall
into two broad categories: those that hydrolyze the glycosides and measure
liberated cyanide and those that extract the intact glycosides and analyze them
chromatographically. Examples of the former category include acid hydrolyzed or
enzyme-linked colorimetric tests (3, 6, 7), and autohydrolysis followed by
quantitation of liberated cyanide by LC (8). Examples of the latter category
include TLC (3, 9–12), RPLC (2, 4, 6, 13–19), and GC (2, 20, 21). Various
extraction methodologies are also found in the literature. The most widely used
extraction method involves extraction of ground seed with 70% methanol in a 30°C
sonicating water bath for 1 h (13, 17). Variations on this method involve
shortening the extraction time to 30 min (4) and using 70% ethanol (6) or 80%
methanol (14, 15) instead of 70% methanol. Other methods include overnight room
temperature extraction of unground seed in methanol on a shaker (2), grinding
in liquid nitrogen followed by extraction with boiling 80% ethanol (3),
grinding to pass a 1 mm sieve and shaking in 0.1 M orthophosphoric acid for 1 h
(22), and electric homogenization in water followed by autohydrolysis (8).
Another method involves extraction of glycosides with 70% ethanol (7°C, 1 h),
evaporation to dryness, resuspension in methanol and then chloroform (1:2
ratio), clarification by centrifugation, evaporation of the supernatnant, and
redissolution of the dried material in 15% aqueous methanol (19).
Only two comparisons between analytical methods
have been published. Haque and Bradbury (7) compared an acid hydrolysis colorimetric
technique to an enzyme-linked colorimetric technique and demonstrated that the
acid hydrolysis method was the less accurate and less reproducible of the two.
Kobaisy et al. (6) demonstrated that two enzyme-linked colorimetric tests were
equally accurate to an RPLC method. Although this establishes the relative
performance of these assays, there is a lack of published data demonstrating
the repeatability, precision, and accuracy of any one method.
Recently, we developed and
validated a GC method for measurement of linustatin and neolinustatin in
flaxseed (20). During development, preliminary

investigations demonstrated that certain factors
have a great effect on extraction efficiency. The choice of grinder affected
the extraction efficiency by up to 12% and the use of three different
extraction methodologies affected extraction efficiency by up to 18%. Given the
variability of other published extraction methodologies, it is likely that the
extraction efficiency would vary even more across all published methods. This
is a major shortcoming, as the results for any one analytical method, no matter
how well-characterized it is, cannot be any better than the extraction method.
This study reports the development of exhaustive and routine extraction
methodology for cyanogenic glycosides from flaxseed.
Materials and Methods
Materials and Reagents
Linustatin and neolinustatin were purchased from
Chromadex Inc. (Santa Ana, CA). Methyl-a-D-glucopyranoside, phenyl-b-D-glucopyranoside,
and 1-methylimidazole were purchased from Sigma-Aldrich (St. Louis, MO).
Bistrimethylsilylacetamide and trimethylsilylchlorosilane (TMCS) were purchased
from Regis Technologies Inc. (Morton Grove, IL). All other solvents were of ACS
grade or better, and purchased from Fisher Scientific (Whitby, ON) or VWR
International (Mississauga, ON), with the exception of anhydrous ethanol, which
was purchased from Commercial Alcohols Inc. (Toronto, ON).
Equipment
The GC apparatus consisted of an
Agilent 6890 gas chromatograph equipped with an Agilent 7683 series
autoinjector (10 mL syringe) and a flame ionization
detector. Data collection and analysis were carried out using Agilent
Chemstation software, Version A.09.03, Build 1417. The column was a Supelco
(St. Louis, MO) SPB-17 column (30 m ´ 0.32 mm,
0.25 mm film thickness). Hydrogen carrier gas was
produced using a Parker (Haverhill, MA) hydrogen generator, Model 75-34. Sample
grinders included an in-house constructed high-speed impact grinder, a Black
and Decker Smartgrind coffee mill, and a Retsch Model ZM200 mill with a 1.0 mm
sieve (Haan, Germany). Extraction equipment included a Tissue Tearor Model
985-370 high-speed electric homogenizer (Biospec Products Inc., Bartlesville,
OK), a Crest Tru-Sweep Model 575HT ultrasonic water bath (Trenton, NJ) and a
vortex mixer (various models). Centrifugation was performed on a Beckman GS-6R
centrifuge (Fullerton, CA) equipped with a GH 3.8 rotor.
Flaxseed Samples
Samples were derived from No. 1 Canada Western
flaxseed collected from the Canadian Grain Commission’s 2005 and 2006 harvest
surveys. The FX2005 and FX2006 composites were composites from 2005 and 2006,
respectively, consisting of a variety of cultivars (food and industrial use)
and growing locations across Western Canada. FX2006 MB Bethune consisted of
seed of the Bethune

cultivar from 72 growing locations in Manitoba
during the 2006 crop year. FX2006 SK Vimy was a composite of seed of the Vimy
cultivar from 57 growing locations across Saskatchewan.
Analytical Method
Preparation of trimethylsilyl
ester (TMS) derivatives and analysis by GC were carried out as previously
described (20). Positive displacement pipets were used to measure extracts at
all points of sample preparation.
General Extraction Method
Samples were ground using either a coffee grinder
or a Retsch mill (1.0 mm sieve, 18 000 rpm). Ground meal samples were blended
manually for 2 min, and replicate samples (0.51 ± 0.1 g) were weighed into 20
mL glass vials. Extraction solvent (5 mL) was added to each vial, and vials
were capped. All samples were briefly agitated immediately
before extraction in an ultrasonic water bath set at 40°C for 30
min. The water bath temperature typically increased to 47–49°C during
this interval. The extraction solvent was turbid and milky in appearance after
extraction. After cooling for 15 min, samples were centrifuged at 1500 ´ g for 10 min. Extracts were analyzed
within 24 h of preparation and stored at –20°C unless otherwise indicated.
Comparison of Extraction Solvents
A single batch of FX2005
composite seed was ground in the Retsch mill and extracted (n = 6 for each treatment) using
combinations of aqueous ethanol or methanol (50, 60, 70, 80, 90, or 100%
alcohol), as described.
Effect of Solvent-Meal Ratio on Extraction
A single bulk sample of FX2006
composite was ground in the coffee mill. Sextuplicate extractions were
performed using the general method with 75% methanol at each of the


following solvent-to-meal ratios: 20:1 (0.25 g ground seed, 5 mL
extraction solvent); 16:1 (0.25 g, 4 mL); 10:1 (0.5 g, 5 mL); 6.67:1 (0.75 g, 5
mL); 5:1 (1 g, 5 mL); and 2.5:1 (2.5 g, 5 mL). The volumes of extract used to
prepare GC samples were varied as follows to ensure that peak areas were within
the calibration curve: 20:1 (200 mL); 16:1 (150 mL); 10:1
(100 mL); 6.67:1 (75 mL); 5:1 (50 mL); and 2.5:1 (25 mL).
Multiple Extraction of Flaxseed Meal
Single, double, triple, quadruple, and quintuple
extractions were compared using FX2005 composite seed. A single batch of seed
was ground in the Retsch mill and extracted (n = 6 for each treatment) with 75% methanol in water using the
general method (10:1 solvent-to-meal ratio). Extracts were pooled for each
sample and filtered through by syringe filter (0.45 mm, 47 mm
diameter, nylon) before analysis.
Effect of Extraction Time on Extraction
Efficiency
A single bulk sample of ground
meal was prepared using the Retsch mill. Extraction samples were weighed using
the general method (n = 6 for each
treatment) and were triple-extracted. The duration of extraction was set at 5,
15, 30, or 60 min per extraction round. Extracts were pooled for each sample
and filtered before analysis as described in the multiple extraction experiment.
Determination of Extraction Efficiencies of
Routine Extraction Methods
Two routine methods were compared to the
established reference method in order to calculate the extraction efficiencies
of each method. The reference method used seed ground in the Retsch mill and
triple extraction with 75% methanol. The routine methods used the coffee mill
to grind samples and a single extraction with either 80% ethanol or 75%
methanol. All three methods used the sonicating water bath, as described in the
general extraction method. Four seed samples were used for comparison: FX2005
composite; FX2006 composite; FX2006 MB Bethune; and FX2006 SK
Vimy. Six extracts were prepared from each meal
sample from each grinder.
Evaluation of Meal and Extract Stability
The stability of ground meal and
extracts was assessed using FX2005 composite seed. Extracts (n = 6 per treatment) were stored at room
temperature, refrigerated temperature (3°C), and
freezer temperature (–18°C) after 1 and 7 days of storage.
Meal stability was assessed at room temperature (2, 4, 6, and 24 h) and at refrigerator
and freezer temperatures (1, 7, 14, and 28 days). Meal samples (n = 6 per treatment) were pre-weighed
into extraction vials with tightly fitting caps and PTFA-lined septa prior to
storage. All meal samples were extracted and analyzed immediately after removal
from storage on the same day along with stored extract samples. All treatment
groups were compared to freshly ground and extracted seed.
Statistical Analysis
All statistical analyses were
performed using SAS 9.1 with Enterprise Guide 4.1. Significant differences
between treatment groups were tested by analysis of variance; when detected,
treatment groups were grouped using the Student-Newman-Keuls (SNK)
multiple-range test. Grubbs’ test for outliers was used for treatments when the
RSD exceeded 7.5%. All tests were performed at the 95% confidence interval.
Results and Discussion
Development of the extraction
methodology was conducted by first selecting an extraction solvent. Combinations
of aqueous ethanol or methanol (50–100%, v/v) were evaluated. Water was also
evaluated; however, extracted mucilage was not soluble in the derivatization
cocktail and rendered the samples non-analyzable. Methanol-containing solvent
systems extracted more linustatin than neolinustatin in the corresponding
amounts of ethanol (Figure 1). The difference between 90 and 100%
ethanol was striking, and demonstrated the importance of the presence of
water in the extraction solvent. The difference between 90 and 100% methanol
showed the same trend. The largest amounts of both linustatin and neolinustatin
were extracted with 70 and 80% methanol. Although the means for 80% methanol
were numerically lower for both species, the difference was not statistically
significant. The 75% methanol was selected as the extraction solvent for
further work to provide a buffer region to control for variability of seed
moisture and extraction solvent batches.
The ratio of solvent volume to mass of ground seed
was set at 10:1 (5 mL to 0.5 g) early in development. To evaluate the
sensitivity of the extraction to this parameter, the ratio was varied as widely
as could reasonably be accommodated by the constraints of the equipment in use:
20:1; 16:1; 10:1; 6.67:1; 5:1; and 2.5:1 (Table 1). Two outliers were
identified using the Grubbs test, one in each of the 20:1 and 6.67:1 treatment
groups. Upon elimination of the outliers, the 20:1, 16:1, 10:1, and 6.67:1
groups were statistically equivalent and showed superior extraction to the 5:1
(which was the same as 6.67:1) and 2.5:1 groups. When the data were tested
without removal of outliers, all ratios were equal except the 2.5:1 group,
which was significantly lower. The solvent-to-ground seed ratio of 10:1 was
maintained for both the routine and reference extraction method.
Extraction efficiency was surprisingly insensitive
to extraction time (Figure 2). There was no difference between 5, 15, 30, or 60
min for linustatin, whereas 5, 30, and 60 min were significantly higher than 15
min for neolinustatin. This difference (15 min being inferior to 5 and 30 min)
is likely anomalous and not an actual relationship, and was likely observed due
to the small variability between replicate extractions within the treatment
group. A 30 min extraction time was selected to provide control for any
foreseeable differences in sonicating water baths between laboratories.
The effect of multiple extraction
was evaluated by subjecting meal samples to up to five replicate extractions (Table 2). Double, triple, quadruple, and quintuple extraction were
statistically equal in efficiency and better than single extraction. The effect
of further particle size reduction (beyond that of the Retsch mill) was
investigated by performing a single extraction with 75% methanol and electric
homogenizer before sonication. Although the yields for linustatin and
neolinustatin were numerically marginally higher than single extraction without
the electric homogenizer, the difference was not significant. Importantly, the
SD value increased dramatically with the incorporation of the extra step. The
use of addition particle reducing steps was not considered any further. Two
candidate routine methods using a coffee mill rather than the Retsch and either
75% methanol (routine methanol method) or 80% ethanol (rapid ethanol method)
were also investigated. The extraction efficiency was expectedly lower for both
methods. When the coffee mill is compared to the Retsch (single extraction, 75%
methanol), the results from the coffee mill are significantly lower than those
from the Retsch mill. This may be attributed directly to the superior ability
of the Retsch (impact plus sieving) mill over the coffee mill (impact only) at
particle size reduction. This difference also illustrates the importance of
adequate and consistent grinding in any extraction method.
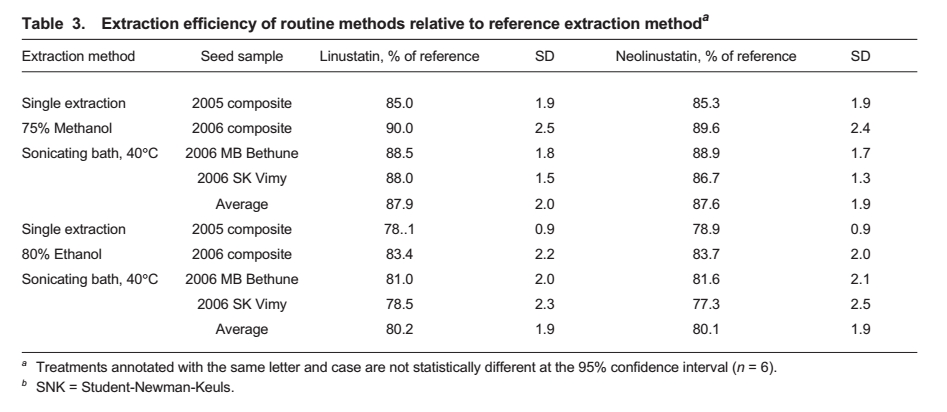
The extraction efficiency of the routine ethanol
and methanol methods was determined using four flax composites (Table 3). The
2005 and 2006 composites represented an average sample matrix that would be
expected from a Canadian sample, whereas the MB Bethune and SK Vimy represented
the two most popular cultivars grown in Canada. Typically, Bethune comprises
over 55% and Vimy 10–15% of total Canadian flax (Canadian Grain Commission).
The mean extraction efficiencies were 87.9 ± 2.0% SD (linustatin) and 87.6 ±
1.9% SD (neolinustatin) for the routine methanol extraction, and 80.2 ± 1.9% SD
(linustatin) and 80.1 ± 1.9% SD (neolinustatin) for the routine ethanol
extraction. Both methods showed low variability in extraction efficiency, but
the routine methanol method was the better choice because of

the higher recovery. The use of a routine
extraction method and extraction efficiency correction increased the overall
measurement uncertainty, compared to using the reference method for every
sample. The choice between routine and reference method should be made by
balancing the need for high throughput versus best possible accuracy.
The establishment of sample stability is critical
to the development of a method, as it defines sample storage conditions,
stability windows and stability of ground meal samples; extracts were assessed
by storage of samples and comparison to freshly ground, freshly extracted
samples (Table 4). Stability of ground meal was assessed at room temperature
over 2, 4, and 6 h periods to simulate laboratory conditions where seed may be
ground several hours before extraction. Meal and extract storage were also
assessed at room temperature, refrigeration, and freezer conditions over 1 and
7 days. Finally, meal was assessed at 2 weeks and at 2 months in the freezer.
The freshly ground and extracted sample yielded the highest assay results for
both analytes and were not statistically different from any storage conditions
other than meal at 1 day under refrigeration (linustatin), extract after 7 days
at room temperature (linustatin), and meal after 2 months in the freezer (both
analytes). The difference observed in the 1 day meal treatment was likely an
anomaly arising from the low SD values observed, as the meal was statistically
equivalent to the fresh sample after 1 and 7 days at room temperature (a
harsher condition), as well as 7 days under refrigeration. The statistically
lower results for the extract after 7 days at room temperature and meal after 2
months in the freezer must be accepted, however, as no longer-term samples were
analyzed. When the statistics were repeated at the 99% confidence interval
(data not shown), all storage conditions were statistically inseparable from
the freshly ground and extracted sample, with the exception of the 2 month meal
sample. It was concluded that ground meal could be stored at room temperature
for at least 24 h and up to a week without analyte degradation, and extracts
may be stored at room temperature for up to 1 day, but should be refrigerated
or frozen thereafter.
In conclusion, the reference
extraction method for linustatin and neolinustatin from flax consisted of
sample grinding using a Retsch ZM200 mill (or equivalent) at 18 000 rpm with a
1.0 mm sieve coupled with triple-pooled extraction in a sonicating water bath
(40°C, 30 min) using 75% methanol. The routine method
differed by the use of a coffee mill to grind samples and a single extraction.
The largest barrier to method transferability is the
lack of a standardized or certified reference material. This means that, due to
differences in grinding equipment and with no reference material for
comparison, conditions of (essentially) complete extraction must be
re-established in every laboratory for the reference extraction method.
Acknowledgments
This is Canadian Grain Commission paper No. 1026.
References
(1)
Smith, C.R., Jr, Weisleider, D.,
& Miller, R.W. (1980) J. Org. Chem. 45, 507–510
(2)
Frehner, M., Scalet, M., &
Conn, E.E. (1990) Plant Physiol. 94, 28–34
(3) Niedïwiedï-Siegie½,
I. (1998) Phytochemistry 49, 59–63
(4)
Oomah, B.D., Mazza, G., &
Naschuk, E.O. (1992) J. Agric. Food Chem. 40, 1346–1348
(5)
Barthet, V.B., & Bacala, R.
(2009) in Compendium of Bioactive Natural Products, Vol. 6, V.K.
Gupta (Ed.), Stadium Press LLC,
Houston, TX
(6)
Kobaisy, M., Oomah, B.D., &
Mazza, G. (1996) J. Agric. Food Chem. 44, 3178–3181
(7)
Haque, M.R., & Bradbury, J.H.
(2002) Food Chem. 77, 107–114
(8)
Chadha, R.K., Lawrence, J.F.,
& Ratnayake, W.M.N. (1995) Food
Addit. Contam. 12, 527–533
(9)
Amarowicz, R., Wanasundara,
P.K.J.P.D., & Shahidi, F. (1993) Die
Nahrung 1, 88–90
(10)
Smith, C.R., Jr, Weisleder, D.,
Miller, R.W., Palmer, I.S., & Olson, O.E. (1980) J. Org. Chem. 45, 507–510
(11)
Palmer, I.S., Olson, O.E.,
Halverson, A.W., Miller, R., & Smith, C. (1980) J. Nutr. 110, 145–150
(12)
Brimer, L., Christensen, S.B., Mrlgaard,
P., & Nartey, F. (1983) J. Agric.
Food Chem. 31, 789–793
(13)
von Schilcher, H., &
Wilkens-Sauter, M. (1986) Fett. Wis. Technol. 88, 287–290
(14)
Krech, M.J., & Fieldes, M.A.
(2003) Can. J. Bot. 81, 1029–1038
(15)
Park, E.R., Hong, J.H., Lee,
D.H., Han, S.B., Lee, K.B., Park, J.S., Chung, H.W., Hong, K.H., & Kim,
M.C. (2005) J. Korean Soc. Food Sci. Nutr. 34,
875–879
(16)
Kolodziejczyk, P.P., & Fedec,
P. (1995) in Flaxseed and Human Nutrition, 1st Ed., S.C. Cunnane
& L.U. Thompson (Eds.), AOCS
Press, Champaign, IL, pp 261–280
(17)
Cunnane, S.C., Ganguli, S.,
Menard, C., Liede, A.C., Hamadeh, M.J., Chen, Z., Wolever, T.M.S., &
Jenkins, D.J.A. (1993) Brit. J. Nutr.
69, 443–453
(18)
Amarowicz, R., Chong, X., &
Shahidi, F. (1993) Food Chem. 48, 99–101
(19)
Wanasundara, P.K.J.P.D.,
Amarowicz, R., Kara, M.T., & Shahidi, F. (1993) Food Chem. 48, 263–266
(20) Bacala,
R., & Barthet, V. (2007) J. AOAC Int.
90, 153–161
(21)
Zilg, H., Tapper, B.A., &
Conn, E.E. (1972) J. Biol. Chem. 247, 2384–2386
(22)
Harris, J.R., Merson, G.H.J.,
Hardy, M.J., & Curtis, D.J. (1980) Analyst
105, 974–980


