Stockley’s Drug Interactions
A source book of interactions, their mechanisms, clinical importance and management
Eighth edition
Edited by Karen Baxter
BSc, MSc, MRPharmS
Contents
Preface v
Abbreviations vi
Before using this book vii
Abbreviations vi
Before using this book vii
1. General considerations and an outline
survey of some basic interaction mechanisms 1
2. ACE inhibitors and Angiotensin II receptor
antagonists 12
3. Alcohol 40
4. Alpha blockers 83
5. Anaesthetics and Neuromuscular blockers 90
6. Analgesics and NSAIDs 133
7. Anorectics and Stimulants 199
8. Anthelmintics, Antifungals and Antiprotozoals 207
9. Antiarrhythmics 243
10. Antibacterials 285
11. Anticholinesterases 352
12. Anticoagulants 358
13. Antidiabetics 468
14. Antiepileptics 517
15. Antihistamines 582
16. Antimigraine drugs 597
17. Antineoplastics 609
18. Antiparkinsonian and related drugs 672
19. Antiplatelet drugs and Thrombolytics 697
20. Antipsychotics, Anxiolytics and Hypnotics 706
21. Antivirals 772
22. Beta blockers 833
23. Calcium-channel blockers 860
24. Cardiovascular drugs, miscellaneous 878
25. Digitalis glycosides 903
26. Diuretics 944
27. Gastrointestinal drugs 960
28. Hormonal contraceptives and Sex hormones 975
29. Immunosuppressants 1009
30. Lipid regulating drugs 1086
31. Lithium 1111
32. MAOIs 1130
33. Respiratory drugs 1158
34. SSRIs, Tricyclics and related antidepressants 1203
35. Miscellaneous drugs 1247
Index 1293
Preface
The aim of Stockley’s Drug Interactions is to inform busy doctors, pharmacists, surgeons, nurses and other healthcare professionals, of the facts about drug interactions, without their having to do the time-consuming literature searches and full assessment of the papers for themselves. These therefore
are the practical questions which this book attempts to answer:
• Are the drugs and substances in question known to interact or is the interaction only theoretical and speculative?
• If they do interact, how serious is it?
• Has it been described many times or only once?
• Are all patients affected or only a few?
• Is it best to avoid these two substances altogether or can the interaction be accommodated in some way?
• And what alternative and safer drugs can be used instead?
To précis the mass of literature into a concise and easy-to-read form, the text has been organised into a series of individual monographs, all with a common format. If you need some insight into the general philosophy underlying the way all this information is handled in this publication, you should have a look at the section, ‘Before using this book. . .’.
There have been several changes for the 8th edition. All of the existing monographs have, as with each edition, been reviewed, revalidated and updated, and many new ones have been added, making a total in excess of 3100 monographs. Many new monographs on herbal interactions have been added, although good quality human studies remain sparse. A new chapter has been added to cover the growing number of interactions about anorectics, and the chapter on sympathomimetics has been removed, with the information redistributed according to the therapeutic use of the drugs in question, to give a better indication of precisely which drugs from this disparate group are likely to interact. We have continued to add information provided by regulatory bodies outside of the UK, which further enhances the international flavour of the publication.
The aim of Stockley’s Drug Interactions is to inform busy doctors, pharmacists, surgeons, nurses and other healthcare professionals, of the facts about drug interactions, without their having to do the time-consuming literature searches and full assessment of the papers for themselves. These therefore
are the practical questions which this book attempts to answer:
• Are the drugs and substances in question known to interact or is the interaction only theoretical and speculative?
• If they do interact, how serious is it?
• Has it been described many times or only once?
• Are all patients affected or only a few?
• Is it best to avoid these two substances altogether or can the interaction be accommodated in some way?
• And what alternative and safer drugs can be used instead?
To précis the mass of literature into a concise and easy-to-read form, the text has been organised into a series of individual monographs, all with a common format. If you need some insight into the general philosophy underlying the way all this information is handled in this publication, you should have a look at the section, ‘Before using this book. . .’.
There have been several changes for the 8th edition. All of the existing monographs have, as with each edition, been reviewed, revalidated and updated, and many new ones have been added, making a total in excess of 3100 monographs. Many new monographs on herbal interactions have been added, although good quality human studies remain sparse. A new chapter has been added to cover the growing number of interactions about anorectics, and the chapter on sympathomimetics has been removed, with the information redistributed according to the therapeutic use of the drugs in question, to give a better indication of precisely which drugs from this disparate group are likely to interact. We have continued to add information provided by regulatory bodies outside of the UK, which further enhances the international flavour of the publication.
This edition has also seen the growth in our editorial team, with two practising clinical pharmacists recruited to help us ensure we maintain the practical nature of the information given. This has also allowed us to develop our product range, with the publication of the first Stockley’s Drug Interactions
Pocket Companion, which we have developed for delivery on PDA.
As always, the Editorial team have had assistance from many other people in developing this publication , and the Editor gratefully acknowledges the assistance and guidance that they have provided. The Martindale team continue to be a great source of advice and support, and particular thanks is due to the editor, Sean Sweetman. Thanks are also due to John Wilson and Tamsin Cousins, who handle the various aspects of producing our publications in print. We are also grateful for the support of both Paul Weller and Charles Fry. Ivan Stockley remains an important part of the publication, taking a keen interest in the development of new products, and as ever, we find his advice invaluable.
Stockley’s Drug Interactions continues to be available on the Pharmaceutical Press platform, MedicinesComplete, as well as being available on other platforms, both in English and Spanish. With the further development of the integratable Alerts product and the new PDA, we remain indebted to
Julie McGlashan, Michael Evans, Elizabeth King, and all those involved in the development of these products, for their advice and support. For more details about these digital products please visit: www.pharmpress.com/Stockley
As ever, we have had feedback from pharmacists and doctors about the content of the publication, which is always valuable, especially in ensuring the publication meets the needs of the users. We are particularly grateful to those who have taken the time to answer our questions about specific aspects of practice. Anyone who wishes to contact the Stockley team can do so at the following address: stockley@rpsgb.org
Pocket Companion, which we have developed for delivery on PDA.
As always, the Editorial team have had assistance from many other people in developing this publication , and the Editor gratefully acknowledges the assistance and guidance that they have provided. The Martindale team continue to be a great source of advice and support, and particular thanks is due to the editor, Sean Sweetman. Thanks are also due to John Wilson and Tamsin Cousins, who handle the various aspects of producing our publications in print. We are also grateful for the support of both Paul Weller and Charles Fry. Ivan Stockley remains an important part of the publication, taking a keen interest in the development of new products, and as ever, we find his advice invaluable.
Stockley’s Drug Interactions continues to be available on the Pharmaceutical Press platform, MedicinesComplete, as well as being available on other platforms, both in English and Spanish. With the further development of the integratable Alerts product and the new PDA, we remain indebted to
Julie McGlashan, Michael Evans, Elizabeth King, and all those involved in the development of these products, for their advice and support. For more details about these digital products please visit: www.pharmpress.com/Stockley
As ever, we have had feedback from pharmacists and doctors about the content of the publication, which is always valuable, especially in ensuring the publication meets the needs of the users. We are particularly grateful to those who have taken the time to answer our questions about specific aspects of practice. Anyone who wishes to contact the Stockley team can do so at the following address: stockley@rpsgb.org
London, September 2007
Abbreviations
ACE—angiotensin-converting enzyme
ADP—adenosine diphosphate
AIDS—acquired immunodeficiency syndrome
ALL—acute lymphoblastic leukaemia
ALT—alanine aminotransferase
am—ante meridiem (before noon)
AML—acute myeloid leukaemia
aPTT—activated partial thromboplastin time
AST—aspartate aminotransferase
AUC—area under the time–concentration curve
AUC0–12—area under the time–concentration curve measured over 0 to
12 hours
AV—atrioventricular
BNF—British National Formulary
BP—blood pressure
BP—British Pharmacopoeia
BPC—British Pharmaceutical Codex
BPH—benign prostatic hyperplasia
bpm—beats per minute
BUN—blood urea nitrogen
CAPD—continuous ambulatory peritoneal dialysis
CDC—Centers for Disease Control (USA)
CNS—central nervous system
COPD—chronic obstructive pulmonary disease
CPR—cardiopulmonary resuscitation
CSF—cerebrospinal fluid
CSM—Committee on Safety of Medicines (UK) (now subsumed within
the Commission on Human Medicines)
DNA—deoxyribonucleic acid
ECG—electrocardiogram
ECT—electroconvulsive therapy
ED50—the dose at which 50% of subjects respond
EEG—electroencephalogram
e.g.—exempli gratia (for example)
EMEA—The European Agency for the Evaluation of Medicinal
Products
FDA—Food and Drug Administration (USA)
FEF25–75—maximum expiratory flow over the middle 50% of the vital
capacity
FEV1—forced expiratory volume in one second
FSH—follicle simulating hormone
ft—foot (feet)
FVC—forced vital capacity
GGT—gamma glutamyl transpeptidase
g—gram(s)
h—hour(s)
HAART—highly active antiretroviral therapy
HCV—hepatitis C virus
HIV —human immunodeficiency virus
HRT—hormone replacement therapy
ibid—ibidem, in the same place (journal or book)
i.e.—id est (that is)
INR—international normalised ratio
ACE—angiotensin-converting enzyme
ADP—adenosine diphosphate
AIDS—acquired immunodeficiency syndrome
ALL—acute lymphoblastic leukaemia
ALT—alanine aminotransferase
am—ante meridiem (before noon)
AML—acute myeloid leukaemia
aPTT—activated partial thromboplastin time
AST—aspartate aminotransferase
AUC—area under the time–concentration curve
AUC0–12—area under the time–concentration curve measured over 0 to
12 hours
AV—atrioventricular
BNF—British National Formulary
BP—blood pressure
BP—British Pharmacopoeia
BPC—British Pharmaceutical Codex
BPH—benign prostatic hyperplasia
bpm—beats per minute
BUN—blood urea nitrogen
CAPD—continuous ambulatory peritoneal dialysis
CDC—Centers for Disease Control (USA)
CNS—central nervous system
COPD—chronic obstructive pulmonary disease
CPR—cardiopulmonary resuscitation
CSF—cerebrospinal fluid
CSM—Committee on Safety of Medicines (UK) (now subsumed within
the Commission on Human Medicines)
DNA—deoxyribonucleic acid
ECG—electrocardiogram
ECT—electroconvulsive therapy
ED50—the dose at which 50% of subjects respond
EEG—electroencephalogram
e.g.—exempli gratia (for example)
EMEA—The European Agency for the Evaluation of Medicinal
Products
FDA—Food and Drug Administration (USA)
FEF25–75—maximum expiratory flow over the middle 50% of the vital
capacity
FEV1—forced expiratory volume in one second
FSH—follicle simulating hormone
ft—foot (feet)
FVC—forced vital capacity
GGT—gamma glutamyl transpeptidase
g—gram(s)
h—hour(s)
HAART—highly active antiretroviral therapy
HCV—hepatitis C virus
HIV —human immunodeficiency virus
HRT—hormone replacement therapy
ibid—ibidem, in the same place (journal or book)
i.e.—id est (that is)
INR—international normalised ratio
ITU—intensive therapy unit
IU—International Units
IUD—intra-uterine device
kg—kilogram(s)
l—litre
lbs—pound(s) avoirdupois
LDL—low-density lipoprotein
LFT—liver function test
LH—luteinising hormone
LMWH—low-molecular-weight heparin
MAC—minimum alveolar concentration
MAOI—monoamine oxidase inhibitor
MAOI-A—monoamine oxidase inhibitor, type A
MAOI-B—monoamine oxidase inhibitor, type B
MCA—Medicines Control Agency (UK) (now MHRA)
MHRA—Medicines and Healthcare products Regulatory Agency (UK)
MIC—minimum inhibitory concentration
mEq—milliequivalent(s)
mg—milligram(s)
mL—millilitre(s)
mmHg—millimetre(s) of mercury
mmol—millimole
mol—mole
MRSA—methicillin resistant Staphylococcus aureus
NICE—National Institue for Health and Clinical Excellence (UK)
(formerly the National Institute for Clinical Excellence)
nM—nanomole
nmol—nanomole
NNRTI—non-nucleoside reverse transcriptase inhibitor
NRTI—nucleoside reverse transcriptase inhibitor
NSAID—non-steroidal anti-inflammatory drug
PABA—para-amino benzoic acid
PCP—pneumocystis pneumonia
pH—the negative logarithm of the hydrogen ion concentration
pm—post meridiem (after noon)
pO2—plasma partial pressure (concentration) of oxygen
PPI—proton pump inhibitor
ppm—parts per million
PTT—partial thromboplastin time
PUD—peptic ulcer disease
RIMA—reversible inhibitor of monoamine oxidase type A
RNA—ribonucleic acid
sic—written exactly as it appears in the original
SNRI—serotonin and noradrenaline reuptake inhibitor
SSRI—selective serotonin reuptake inhibitor
STD—sexually transmitted disease
SVT—supraventricular tachycardia
TPN—total parenteral nutrition
TSH—thyroid-stimulating hormone
UK—United Kingdom
US and USA—United States of America
USP—The United States Pharmacopeia
UTI—urinary tract infection
IU—International Units
IUD—intra-uterine device
kg—kilogram(s)
l—litre
lbs—pound(s) avoirdupois
LDL—low-density lipoprotein
LFT—liver function test
LH—luteinising hormone
LMWH—low-molecular-weight heparin
MAC—minimum alveolar concentration
MAOI—monoamine oxidase inhibitor
MAOI-A—monoamine oxidase inhibitor, type A
MAOI-B—monoamine oxidase inhibitor, type B
MCA—Medicines Control Agency (UK) (now MHRA)
MHRA—Medicines and Healthcare products Regulatory Agency (UK)
MIC—minimum inhibitory concentration
mEq—milliequivalent(s)
mg—milligram(s)
mL—millilitre(s)
mmHg—millimetre(s) of mercury
mmol—millimole
mol—mole
MRSA—methicillin resistant Staphylococcus aureus
NICE—National Institue for Health and Clinical Excellence (UK)
(formerly the National Institute for Clinical Excellence)
nM—nanomole
nmol—nanomole
NNRTI—non-nucleoside reverse transcriptase inhibitor
NRTI—nucleoside reverse transcriptase inhibitor
NSAID—non-steroidal anti-inflammatory drug
PABA—para-amino benzoic acid
PCP—pneumocystis pneumonia
pH—the negative logarithm of the hydrogen ion concentration
pm—post meridiem (after noon)
pO2—plasma partial pressure (concentration) of oxygen
PPI—proton pump inhibitor
ppm—parts per million
PTT—partial thromboplastin time
PUD—peptic ulcer disease
RIMA—reversible inhibitor of monoamine oxidase type A
RNA—ribonucleic acid
sic—written exactly as it appears in the original
SNRI—serotonin and noradrenaline reuptake inhibitor
SSRI—selective serotonin reuptake inhibitor
STD—sexually transmitted disease
SVT—supraventricular tachycardia
TPN—total parenteral nutrition
TSH—thyroid-stimulating hormone
UK—United Kingdom
US and USA—United States of America
USP—The United States Pharmacopeia
UTI—urinary tract infection
Before using this book . . .
. . . you should read this short explanatory section so that you know how the drug interaction data have been set out here, and why – as well as the basic philosopy that has been followed in presenting it.
The monographs
This publication has over 3100 monographs with a common format, which are subdivided into sections like these:
• An abstract or summary for quick reading.
• Clinical evidence, detailing one, two or more illustrative examples of the interaction, followed by most or all of other supportive clinical evidence currently available.
• Mechanism, in brief.
• Importance and management, a short discussion designed to aid rapid clinical decision making. For example:
– Is the interaction established or not?
– What is its incidence?
– How important is it?
– How can it be managed?
– And what, if any, are the non-interacting alternatives?
• References, a list of all of the relevant references. The length of the references list gives a very fair indication of the extent of the documentation.
. . . you should read this short explanatory section so that you know how the drug interaction data have been set out here, and why – as well as the basic philosopy that has been followed in presenting it.
The monographs
This publication has over 3100 monographs with a common format, which are subdivided into sections like these:
• An abstract or summary for quick reading.
• Clinical evidence, detailing one, two or more illustrative examples of the interaction, followed by most or all of other supportive clinical evidence currently available.
• Mechanism, in brief.
• Importance and management, a short discussion designed to aid rapid clinical decision making. For example:
– Is the interaction established or not?
– What is its incidence?
– How important is it?
– How can it be managed?
– And what, if any, are the non-interacting alternatives?
• References, a list of all of the relevant references. The length of the references list gives a very fair indication of the extent of the documentation.
A long list indicates a well documented interaction, whereas a short list indicates poor documentation.
Some of the monographs have been compressed into fewer subsections instead of the more usual five, simply where information is limited or where there is little need to be more expansive.
The monographs do not carry the drug interaction Hazard/Severity ratings as used in the electronic Stockley Interactions Alerts because of the difficulties of applying them to monographs that cover multiple pairs of drug–drug interactions, but what is written in each monograph should speak for itself.
Some of the monographs have been compressed into fewer subsections instead of the more usual five, simply where information is limited or where there is little need to be more expansive.
The monographs do not carry the drug interaction Hazard/Severity ratings as used in the electronic Stockley Interactions Alerts because of the difficulties of applying them to monographs that cover multiple pairs of drug–drug interactions, but what is written in each monograph should speak for itself.
Quality of information on interactions
The data on interactions are of widely varying quality and reliability.
The best come from clinical studies carried out on large numbers of patients under scrupulously controlled conditions. The worst are anecdotal, uncontrolled, or based solely on animal studies. Sometimes they are no more than speculative and theoretical scaremongering guesswork, hallowed by repeated quotation until they become virtually set in stone.
The aim has been to filter out as much useless noise as possible, so wherever possible ‘secondary’ references are avoided, and ‘primary’ references which are available in good medical and scientific libraries are used instead – although sometimes unpublished, good quality, in-house reports on drug company files have been used where the drug company has kindly allowed access to the information. Product literature (the Summary of Product Characteristics in the UK and the Prescribing Information
in the US) rather than the research reports that lie behind them are also cited because they are the only source of published information about new drugs.
The quality of drug company literature is very variable. Some of it is excellent, helpful and very reliable, but regrettably a growing proportion contains a welter of speculative and self-protective statements, probably driven more by the company's medico-legal policy than anything else, and the nervousness of drug regulatory authorities. It is almost unbelievable (but true all the same) that drug companies that are scrupulous in the way they do their research, come out with statements about possible interactions that are little more than guesswork.
When drawing your own conclusions
The human population is a total mixture, unlike selected batches of laboratory animals (same age, weight, sex, and strain etc.). For this reason human beings do not respond uniformly to one or more drugs. Our genetic make up, ethnic background, sex, renal and hepatic functions, diseases and nutritional states, ages and other factors (the route of administration, for example) all contribute towards the heterogeneity of our responses.
This means that the outcome of giving one or more drugs to any individual for the first time is never totally predictable because it is a new and unique ‘experiment’. Even so, some idea of the probable outcome of using a drug or a pair of drugs can be based on what has been seen in other patients: the more extensive the data, the firmer the predictions.
The most difficult decisions concern isolated cases of interaction, many of which only achieved prominence because they were serious. Do you ignore them as ‘idiosyncratic’ or do you, from that moment onwards, contraindicate the use of the two drugs totally?
There is no simple ‘yes’ or ‘no’ answer to these questions, but one simple rule-of-thumb is that isolated cases of interaction with old and very well-tried pairs of drugs are unlikely to be of general importance, whereas those with new drugs may possibly be the tip of an emerging iceberg and should therefore initially be taken much more seriously until more is known. The delicate balance between these two has then to be set against the actual severity of the reaction reported and weighed up against how essential it is to use the drug combination in question.
When deciding the possible first-time use of any two drugs in any particular patient, you need to put what is currently known about these drugs against the particular profile of your patient. Read the monograph. Consider the facts and conclusions, and then set the whole against the backdrop of your patients unique condition (age, disease, general condition, and so forth) so that what you eventually decide to do is well thought out and soundly based. We do not usually have the luxury of knowing absolutely all the facts, so that an initial conservative approach is often the safest.
1
The human population is a total mixture, unlike selected batches of laboratory animals (same age, weight, sex, and strain etc.). For this reason human beings do not respond uniformly to one or more drugs. Our genetic make up, ethnic background, sex, renal and hepatic functions, diseases and nutritional states, ages and other factors (the route of administration, for example) all contribute towards the heterogeneity of our responses.
This means that the outcome of giving one or more drugs to any individual for the first time is never totally predictable because it is a new and unique ‘experiment’. Even so, some idea of the probable outcome of using a drug or a pair of drugs can be based on what has been seen in other patients: the more extensive the data, the firmer the predictions.
The most difficult decisions concern isolated cases of interaction, many of which only achieved prominence because they were serious. Do you ignore them as ‘idiosyncratic’ or do you, from that moment onwards, contraindicate the use of the two drugs totally?
There is no simple ‘yes’ or ‘no’ answer to these questions, but one simple rule-of-thumb is that isolated cases of interaction with old and very well-tried pairs of drugs are unlikely to be of general importance, whereas those with new drugs may possibly be the tip of an emerging iceberg and should therefore initially be taken much more seriously until more is known. The delicate balance between these two has then to be set against the actual severity of the reaction reported and weighed up against how essential it is to use the drug combination in question.
When deciding the possible first-time use of any two drugs in any particular patient, you need to put what is currently known about these drugs against the particular profile of your patient. Read the monograph. Consider the facts and conclusions, and then set the whole against the backdrop of your patients unique condition (age, disease, general condition, and so forth) so that what you eventually decide to do is well thought out and soundly based. We do not usually have the luxury of knowing absolutely all the facts, so that an initial conservative approach is often the safest.
1
General considerations and an outline survey of some basic interaction mechanisms
A. What is a drug interaction?
An interaction is said to occur when the effects of one drug are changed by the presence of another drug, herbal medicine, food, drink or by some environmental chemical agent. Much more colourful and informal definitions by patients are that it is “. . . when medicines fight each other. . .”, or“. . . when medicines fizz together in the stomach . . .”, or “. . .what happens when one medicine falls out with another. . .”
The outcome can be harmful if the interaction causes an increase in the toxicity of the drug. For example, there is a considerable increase in risk of severe muscle damage if patients on statins start taking azole antifungals (see ‘Statins + Azoles’, p.1093).
The outcome can be harmful if the interaction causes an increase in the toxicity of the drug. For example, there is a considerable increase in risk of severe muscle damage if patients on statins start taking azole antifungals (see ‘Statins + Azoles’, p.1093).
Patients taking monoamine oxidase inhibitor antidepressants (MAOIs) may experience an acute and potentially life-threatening hypertensive crisis if they eat tyramine-rich foods such
as ‘cheese’, (p.1153).
A reduction in efficacy due to an interaction can sometimes be just as harmful as an increase: patients taking warfarin who are given rifampicin need more warfarin to maintain adequate and protective anticoagulation (see ‘Coumarins + Antibacterials; Rifamycins’, p.375), while patients taking ‘tetracyclines’, (p.347) or ‘quinolones’, (p.332) need to avoid antacids and milky foods (or separate their ingestion) because the effects of these antibacterials can be reduced or even abolished if admixture occurs in the gut.
These unwanted and unsought-for interactions are adverse and undesirable but there are other interactions that can be beneficial and valuable, such as the deliberate co-prescription of antihypertensive drugs and diuretics in order to achieve antihypertensive effects possibly not obtainable with either drug alone. The mechanisms of both types of interaction, whether adverse or beneficial, are often very similar, but the adverse interactions are the focus of this publication.
Definitions of a drug interaction are not rigidly adhered to in this publication because the subject inevitably overlaps into other areas of adverse reactions with drugs. So you will find in these pages some ‘interactions’ where one drug does not actually affect another at all, but the adverse outcome is the simple additive effects of two drugs with similar effects (for example the combined effects of two or more CNS depressants, or two drugs which affect the QT interval). Sometimes the term ‘drug interaction’ is used for the physico-chemical reactions that occur if drugs are mixed in intravenous fluids, causing precipitation or inactivation.
as ‘cheese’, (p.1153).
A reduction in efficacy due to an interaction can sometimes be just as harmful as an increase: patients taking warfarin who are given rifampicin need more warfarin to maintain adequate and protective anticoagulation (see ‘Coumarins + Antibacterials; Rifamycins’, p.375), while patients taking ‘tetracyclines’, (p.347) or ‘quinolones’, (p.332) need to avoid antacids and milky foods (or separate their ingestion) because the effects of these antibacterials can be reduced or even abolished if admixture occurs in the gut.
These unwanted and unsought-for interactions are adverse and undesirable but there are other interactions that can be beneficial and valuable, such as the deliberate co-prescription of antihypertensive drugs and diuretics in order to achieve antihypertensive effects possibly not obtainable with either drug alone. The mechanisms of both types of interaction, whether adverse or beneficial, are often very similar, but the adverse interactions are the focus of this publication.
Definitions of a drug interaction are not rigidly adhered to in this publication because the subject inevitably overlaps into other areas of adverse reactions with drugs. So you will find in these pages some ‘interactions’ where one drug does not actually affect another at all, but the adverse outcome is the simple additive effects of two drugs with similar effects (for example the combined effects of two or more CNS depressants, or two drugs which affect the QT interval). Sometimes the term ‘drug interaction’ is used for the physico-chemical reactions that occur if drugs are mixed in intravenous fluids, causing precipitation or inactivation.
The long-established and less ambiguous term is ‘pharmaceutical incompatibilities’.
Incompatibilities are not covered by this publication.
Incompatibilities are not covered by this publication.
B. What is the incidence of drug interactions?
The more drugs a patient takes the greater the likelihood that an adverse reaction will occur. One hospital study found that the rate was 7% in those taking 6 to 10 drugs but 40% in those taking 16 to 20 drugs, which represents a disproportionate increase.1 A possible explanation is that the drugs were interacting.
Some of the early studies on the frequency of interactions uncritically compared the drugs that had been prescribed with lists of possible drug interactions, without appreciating that many interactions may be clinically trivial or simply theoretical. As a result, an unrealistically high incidence was suggested. Most of the later studies have avoided this error by looking at only potentially clinically important interactions, and incidences of up to 8.8% have been reported.2-4 Even so, not all of these studies took into account the distinction that must be made between the incidence of potential interactions and the incidence of those where clinical problems actually arise. The simple fact is that some patients experience quite serious reactions while taking interacting drugs, while others appear not to be affected at all.
A screening of 2 422 patients over a total of 25 005 days revealed that 113 (4.7%) were taking combinations of drugs that could interact, but evidence of interactions was observed in only seven patients, representing only 0.3%.2 In another hospital study of 44 patients over a 5-day period taking 10 to 17 drugs, 77 potential drug interactions were identified, but only one probable and four possible adverse reactions (6.4%) were detected. 5 A further study among patients taking anticonvulsant drugs found that 6% of the cases of toxicity were due to drug interactions.6 These figures are low compared with those of a hospital survey that monitored 927 patients who had received 1004 potentially interacting drug combinations. Changes in drug dosage were made in 44% of these cases.7 A review of these and other studies found that the reported incidence rates ranged from 2.2 to 70.3%, and the percentage of patients actually experiencing problems was less than 11.1%. Another review found a 37% incidence of interactions among 639 elderly patients.8 Yet another review of 236 geriatric patients found an 88% incidence of clinically significant interactions, and a 22% incidence of potentially serious and life-threatening interactions. 9A 4.1% incidence of drug interactions on prescriptions presented to community pharmacists in the USA was found in a further survey,10 whereas the incidence was only 2.9% in another American study,11 and just 1.9% in a Swedish study.12 An Australian study found that about 10% of hospital admissions were drug-related, of which 4.4% were due to drug interactions. 13 A very high incidence (47 to 50%) of potential drug interactions was found in a study carried out in an Emergency Department in the US. 14 One French study found that 16% of the prescriptions for a group of patients taking antihypertensive drugs were contraindicated or unsuitable, 15 whereas another study on a group of geriatrics found only a 1% incidence. 16 The incidence of problems would be expected to be higher in the elderly because ageing affects the functioning of the kidneys and liver.17,18
Some of the early studies on the frequency of interactions uncritically compared the drugs that had been prescribed with lists of possible drug interactions, without appreciating that many interactions may be clinically trivial or simply theoretical. As a result, an unrealistically high incidence was suggested. Most of the later studies have avoided this error by looking at only potentially clinically important interactions, and incidences of up to 8.8% have been reported.2-4 Even so, not all of these studies took into account the distinction that must be made between the incidence of potential interactions and the incidence of those where clinical problems actually arise. The simple fact is that some patients experience quite serious reactions while taking interacting drugs, while others appear not to be affected at all.
A screening of 2 422 patients over a total of 25 005 days revealed that 113 (4.7%) were taking combinations of drugs that could interact, but evidence of interactions was observed in only seven patients, representing only 0.3%.2 In another hospital study of 44 patients over a 5-day period taking 10 to 17 drugs, 77 potential drug interactions were identified, but only one probable and four possible adverse reactions (6.4%) were detected. 5 A further study among patients taking anticonvulsant drugs found that 6% of the cases of toxicity were due to drug interactions.6 These figures are low compared with those of a hospital survey that monitored 927 patients who had received 1004 potentially interacting drug combinations. Changes in drug dosage were made in 44% of these cases.7 A review of these and other studies found that the reported incidence rates ranged from 2.2 to 70.3%, and the percentage of patients actually experiencing problems was less than 11.1%. Another review found a 37% incidence of interactions among 639 elderly patients.8 Yet another review of 236 geriatric patients found an 88% incidence of clinically significant interactions, and a 22% incidence of potentially serious and life-threatening interactions. 9A 4.1% incidence of drug interactions on prescriptions presented to community pharmacists in the USA was found in a further survey,10 whereas the incidence was only 2.9% in another American study,11 and just 1.9% in a Swedish study.12 An Australian study found that about 10% of hospital admissions were drug-related, of which 4.4% were due to drug interactions. 13 A very high incidence (47 to 50%) of potential drug interactions was found in a study carried out in an Emergency Department in the US. 14 One French study found that 16% of the prescriptions for a group of patients taking antihypertensive drugs were contraindicated or unsuitable, 15 whereas another study on a group of geriatrics found only a 1% incidence. 16 The incidence of problems would be expected to be higher in the elderly because ageing affects the functioning of the kidneys and liver.17,18
These discordant figures need to be put into the context of the under-reporting of adverse reactions of any kind by medical professionals, for reasons that may include pressure of work or the fear of litigation. Both doctors and patients may not recognise adverse reactions and interactions, and some patients simply stop taking their drugs without saying why. None of these studies give a clear answer to the question of how frequently drug interactions occur, but even if the incidence is as low as some of the studies suggest, it still represents a very considerable number of patients who appear to be at risk when one thinks of the large numbers of drugs prescribed and taken every day.
1. Smith JW, Seidl LG, Cluff LE. Studies on the epidemiology of adverse drug reactions. V. Clinical factors influencing susceptibility. Ann Intern Med (1969) 65, 629.
2. Puckett WH, Visconti JA. An epidemiological study of the clinical significance of drug-drug interaction in a private community hospital. Am J Hosp Pharm (1971) 28, 247.
3. Shinn AF, Shrewsbury RP, Anderson KW. Development of a computerized drug interaction database (Medicom) for use in a patient specific environment. Drug Inf J (1983) 17, 205.
4. Ishikura C, Ishizuka H. Evaluation of a computerized drug interaction checking system. Int J Biomed Comput (1983) 14, 311.
5. Schuster BG, Fleckenstein L, Wilson JP, Peck CC. Low incidence of adverse reactions due to drug-drug interaction in a potentially high risk population of medical inpatients. Clin Res (1982) 30, 258A.
6. Manon-Espaillat R, Burnstine TH, Remler B, Reed RC, Osorio I. Antiepileptic drug intoxication: factors and their significance. Epilepsia (1991) 32, 96–100.
7. Haumschild MJ, Ward ES, Bishop JM, Haumschild MS. Pharmacy-based computer system for monitoring and reporting drug interactions. Am J Hosp Pharm (1987) 44, 345.
8. Manchon ND, Bercoff E, Lamarchand P, Chassagne P, Senant J, Bourreille J. Fréquence et gravité des interaction médicamenteuses dans une population âgée: étude prospective concernant 639 malades. Rev Med Interne (1989) 10, 521–5.
9. Lipton HL, Bero LA, Bird JA, McPhee SJ. The impact of clinical pharmacists’ consultations on physicians’ geriatric drug prescribing. Med Care (1992) 30, 646–58.
2. Puckett WH, Visconti JA. An epidemiological study of the clinical significance of drug-drug interaction in a private community hospital. Am J Hosp Pharm (1971) 28, 247.
3. Shinn AF, Shrewsbury RP, Anderson KW. Development of a computerized drug interaction database (Medicom) for use in a patient specific environment. Drug Inf J (1983) 17, 205.
4. Ishikura C, Ishizuka H. Evaluation of a computerized drug interaction checking system. Int J Biomed Comput (1983) 14, 311.
5. Schuster BG, Fleckenstein L, Wilson JP, Peck CC. Low incidence of adverse reactions due to drug-drug interaction in a potentially high risk population of medical inpatients. Clin Res (1982) 30, 258A.
6. Manon-Espaillat R, Burnstine TH, Remler B, Reed RC, Osorio I. Antiepileptic drug intoxication: factors and their significance. Epilepsia (1991) 32, 96–100.
7. Haumschild MJ, Ward ES, Bishop JM, Haumschild MS. Pharmacy-based computer system for monitoring and reporting drug interactions. Am J Hosp Pharm (1987) 44, 345.
8. Manchon ND, Bercoff E, Lamarchand P, Chassagne P, Senant J, Bourreille J. Fréquence et gravité des interaction médicamenteuses dans une population âgée: étude prospective concernant 639 malades. Rev Med Interne (1989) 10, 521–5.
9. Lipton HL, Bero LA, Bird JA, McPhee SJ. The impact of clinical pharmacists’ consultations on physicians’ geriatric drug prescribing. Med Care (1992) 30, 646–58.
10. Rupp MT, De Young M, Schondelmeyer SW. Prescribing problems and pharmacist interventions in community practice. Med Care (1992) 30, 926–40.
11. Rotman BL, Sullivan AN, McDonald T, DeSmedt P, Goodnature D, Higgins M, Suermond HJ, Young CY, Owens DK. A randomized evaluation of a computer-based physician’s workstation; design considerations and baseline results. Proc Annu Symp Comput Appl Med Care (1995) 693–7.
12. Linnarsson R. Drug interactions in primary health care. A retrospective database study and its implications for the design of a computerized decision support system. Scand J Prim Health Care (1993) 11, 181–6.
13. Stanton LA, Peterson GM, Rumble RH, Cooper GM, Polack AE. Drug-related admissions to an Australian hospital. J Clin Pharm Ther (1994) 19, 341–7.
14. Goldberg RM, Mabee J, Chan L, Wong S. Drug-drug and drug-disease interactions in the ED; analysis of a high-risk population. Am J Emerg Med (1996) 14, 447–50.
15. Paille R, Pissochet P. L’ordonnance et les interactions medicamenteuses: etude prospective chez 896 patients traites pour hypertension arterielle en medicine generale. Therapie (1995) 50, 253–8.
16. Di Castri A, Jacquot JM, Hemmi P, Moati L, Rouy JM, Compan B, Nachar H, Bossy-Vassal A. Interactions medicamenteuses: etude de 409 ordannances etablies a l’issue d’une hospitalisation geriatrique. Therapie (1995) 50, 259–64.
17. Cadieux RJ. Drug interactions in the elderly. Postgrad Med (1989) 86, 179–86.
18. Tinawi M, Alguire P. The prevalence of drug interactions in hospitalized patients. Clin Res (1992) 40, 773A.
11. Rotman BL, Sullivan AN, McDonald T, DeSmedt P, Goodnature D, Higgins M, Suermond HJ, Young CY, Owens DK. A randomized evaluation of a computer-based physician’s workstation; design considerations and baseline results. Proc Annu Symp Comput Appl Med Care (1995) 693–7.
12. Linnarsson R. Drug interactions in primary health care. A retrospective database study and its implications for the design of a computerized decision support system. Scand J Prim Health Care (1993) 11, 181–6.
13. Stanton LA, Peterson GM, Rumble RH, Cooper GM, Polack AE. Drug-related admissions to an Australian hospital. J Clin Pharm Ther (1994) 19, 341–7.
14. Goldberg RM, Mabee J, Chan L, Wong S. Drug-drug and drug-disease interactions in the ED; analysis of a high-risk population. Am J Emerg Med (1996) 14, 447–50.
15. Paille R, Pissochet P. L’ordonnance et les interactions medicamenteuses: etude prospective chez 896 patients traites pour hypertension arterielle en medicine generale. Therapie (1995) 50, 253–8.
16. Di Castri A, Jacquot JM, Hemmi P, Moati L, Rouy JM, Compan B, Nachar H, Bossy-Vassal A. Interactions medicamenteuses: etude de 409 ordannances etablies a l’issue d’une hospitalisation geriatrique. Therapie (1995) 50, 259–64.
17. Cadieux RJ. Drug interactions in the elderly. Postgrad Med (1989) 86, 179–86.
18. Tinawi M, Alguire P. The prevalence of drug interactions in hospitalized patients. Clin Res (1992) 40, 773A.
C. How seriously should interactions be regarded and handled?
It would be very easy to conclude after browsing through this publication that it is extremely risky to treat patients with more than one drug at a time, but this would be an over-reaction. The figures quoted in the previous section illustrate that many drugs known to interact in some patients, simply fail to do so in others. This partially explains why some quite important drug interactions remained virtually unnoticed for many years, a good example of this being the increase in serum digoxin levels seen with quinidine (see ‘Digitalis glycosides + Quinidine’, p.936).
Examples of this kind suggest that patients apparently tolerate adverse interactions remarkably well, and that many experienced physicians accommodate the effects (such as rises or falls in serum drug levels) without consciously recognising that what they are seeing is the result of an interaction.
One of the reasons it is often difficult to detect an interaction is that, as already mentioned, patient variability is considerable. We now know many of the predisposing and protective factors that determine whether or not an interaction occurs but in practice it is still very difficult to predict what will happen when an individual patient is given two potentially interacting drugs. An easy solution to this practical problem is to choose a noninteracting alternative, but if none is available, it is frequently possible to give interacting drugs together if appropriate precautions are taken. If the effects of the interaction are well-monitored they can often be allowed for, often simply by adjusting the dosages. Many interactions are dose-related so that if the dosage of the causative drug is reduced, the effects on the other drug will be reduced accordingly. Thus a non-prescription dosage of cimetidine may fail to inhibit the metabolism of phenytoin, whereas a larger dose may clearly increase phenytoin levels (see ‘Phenytoin + H2-receptor antagonists’, p.559).
The dosage of the affected drug may also be critical. For example, isoniazid causes the levels of phenytoin to rise, particularly in those individuals who are slow acetylators of isoniazid, and levels may become toxic. If the serum phenytoin levels are monitored and its dosage reduced appropriately, the concentrations can be kept within the therapeutic range (see ‘Phenytoin + Antimycobacterials’, p.550). Some interactions can be accommodated by using another member of the same group of drugs. For example, the serum levels of doxycycline can become subtherapeutic if phenytoin, barbiturates or carbamazepine are given, but other ‘tetracyclines’ (p.346) do not seem to be affected. Erythromycin causes serum lovastatin levels to rise because it inhibits its metabolism, but does not affect pravastatin levels because these two statins are metabolised in different ways (see ‘Statins’, (p.1086)). It is therefore clearly important not to uncritically extrapolate the interactions seen with one drug to all members of the same group.
It is interesting to note in this context that a study in two hospitals in Maryland, USA, found that when interacting drugs were given with warfarin (but not theophylline) the length of hospital stay increased by a little over 3 days, with a rise in general costs because of the need to do more tests to get the balance right.1 So it may be easier, quicker and cheaper to use a non-interacting alternative drug (always provided that its price is not markedly greater).
The variability in patient response has lead to some extreme responses among prescribers. Some clinicians have become over-anxious about interactions so that their patients are denied useful drugs that they might reasonably be given if appropriate precautions are taken. This attitude is exacerbated by some of the more alarmist lists and charts of interactions, which fail to make a distinction between interactions that are very well documented and well established, and those that have only been encountered in a single patient, and which in the final analysis are probably totally idiosyncratic. ‘One swallow does not make a summer’, nor does a serious reaction in a single patient mean that the drugs in question should never again be given to anyone else.
At the other extreme, there are some health professionals who, possibly because they have personally encountered few interactions, fail to consider drug interactions, so that some of their patients are potentially put at risk. An example of this is the fact that cisapride continued to be prescribed with known interacting drugs, even after the rare risk of fatal torsade de pointes arrhythmias, which can cause sudden death, was well established2 (see ‘Cisapride + Miscellaneous’, p.963). The responsible position lies between these two extremes, because a very substantial number of interacting drugs can be given together safely, if the appropriate precautions are taken. There are relatively few pairs of drugs that should always be avoided.
1. Jankel CA, McMillan JA, Martin BC. Effect of drug interactions on outcomes of patient receiving warfarin or theophylline. Am J Hosp Pharm (1994) 51, 661–6.
2. Smalley W, Shatin D, Wysowski DK, Gurwitz J, Andrade SE, Goodman M, Chan KA, Platt R, Schech SD, Ray WA. Contraindicated use of cisapride: impact of food and drug administration regulatory action. JAMA (2000) 284, 3036–9.
Examples of this kind suggest that patients apparently tolerate adverse interactions remarkably well, and that many experienced physicians accommodate the effects (such as rises or falls in serum drug levels) without consciously recognising that what they are seeing is the result of an interaction.
One of the reasons it is often difficult to detect an interaction is that, as already mentioned, patient variability is considerable. We now know many of the predisposing and protective factors that determine whether or not an interaction occurs but in practice it is still very difficult to predict what will happen when an individual patient is given two potentially interacting drugs. An easy solution to this practical problem is to choose a noninteracting alternative, but if none is available, it is frequently possible to give interacting drugs together if appropriate precautions are taken. If the effects of the interaction are well-monitored they can often be allowed for, often simply by adjusting the dosages. Many interactions are dose-related so that if the dosage of the causative drug is reduced, the effects on the other drug will be reduced accordingly. Thus a non-prescription dosage of cimetidine may fail to inhibit the metabolism of phenytoin, whereas a larger dose may clearly increase phenytoin levels (see ‘Phenytoin + H2-receptor antagonists’, p.559).
The dosage of the affected drug may also be critical. For example, isoniazid causes the levels of phenytoin to rise, particularly in those individuals who are slow acetylators of isoniazid, and levels may become toxic. If the serum phenytoin levels are monitored and its dosage reduced appropriately, the concentrations can be kept within the therapeutic range (see ‘Phenytoin + Antimycobacterials’, p.550). Some interactions can be accommodated by using another member of the same group of drugs. For example, the serum levels of doxycycline can become subtherapeutic if phenytoin, barbiturates or carbamazepine are given, but other ‘tetracyclines’ (p.346) do not seem to be affected. Erythromycin causes serum lovastatin levels to rise because it inhibits its metabolism, but does not affect pravastatin levels because these two statins are metabolised in different ways (see ‘Statins’, (p.1086)). It is therefore clearly important not to uncritically extrapolate the interactions seen with one drug to all members of the same group.
It is interesting to note in this context that a study in two hospitals in Maryland, USA, found that when interacting drugs were given with warfarin (but not theophylline) the length of hospital stay increased by a little over 3 days, with a rise in general costs because of the need to do more tests to get the balance right.1 So it may be easier, quicker and cheaper to use a non-interacting alternative drug (always provided that its price is not markedly greater).
The variability in patient response has lead to some extreme responses among prescribers. Some clinicians have become over-anxious about interactions so that their patients are denied useful drugs that they might reasonably be given if appropriate precautions are taken. This attitude is exacerbated by some of the more alarmist lists and charts of interactions, which fail to make a distinction between interactions that are very well documented and well established, and those that have only been encountered in a single patient, and which in the final analysis are probably totally idiosyncratic. ‘One swallow does not make a summer’, nor does a serious reaction in a single patient mean that the drugs in question should never again be given to anyone else.
At the other extreme, there are some health professionals who, possibly because they have personally encountered few interactions, fail to consider drug interactions, so that some of their patients are potentially put at risk. An example of this is the fact that cisapride continued to be prescribed with known interacting drugs, even after the rare risk of fatal torsade de pointes arrhythmias, which can cause sudden death, was well established2 (see ‘Cisapride + Miscellaneous’, p.963). The responsible position lies between these two extremes, because a very substantial number of interacting drugs can be given together safely, if the appropriate precautions are taken. There are relatively few pairs of drugs that should always be avoided.
1. Jankel CA, McMillan JA, Martin BC. Effect of drug interactions on outcomes of patient receiving warfarin or theophylline. Am J Hosp Pharm (1994) 51, 661–6.
2. Smalley W, Shatin D, Wysowski DK, Gurwitz J, Andrade SE, Goodman M, Chan KA, Platt R, Schech SD, Ray WA. Contraindicated use of cisapride: impact of food and drug administration regulatory action. JAMA (2000) 284, 3036–9.

D. Mechanisms of drug interaction
Some drugs interact together in totally unique ways, but as the many examples in this publication amply illustrate, there are certain mechanisms of interaction that are encountered time and time again. Some of these common mechanisms are discussed here in greater detail than space will allow in the individual monographs, so that only the briefest reference need be made there.
Mechanisms that are unusual or peculiar to particular pairs of drugs are detailed within the monographs. Very many drugs that interact do so, not by a single mechanism, but often by two or more mechanisms acting in concert, although for clarity most of the mechanisms are dealt with here as though they occur in isolation. For convenience, the mechanisms of interactions can be subdivided into those that involve the pharmacokinetics of a drug, and those that are pharmacodynamic.
Mechanisms that are unusual or peculiar to particular pairs of drugs are detailed within the monographs. Very many drugs that interact do so, not by a single mechanism, but often by two or more mechanisms acting in concert, although for clarity most of the mechanisms are dealt with here as though they occur in isolation. For convenience, the mechanisms of interactions can be subdivided into those that involve the pharmacokinetics of a drug, and those that are pharmacodynamic.
1. Pharmacokinetic interactions
Pharmacokinetic interactions are those that can affect the processes by which drugs are absorbed, distributed, metabolised and excreted (the socalled ADME interactions).
1.1. Drug absorption interactions
Most drugs are given orally for absorption through the mucous membranes of the gastrointestinal tract, and the majority of interactions that go on within the gut result in reduced rather than increased absorption. A clear distinction must be made between those that decrease the rate of absorption and those that alter the total amount absorbed. For drugs that are given long-term, in multiple doses (e.g. the oral anticoagulants) the rate of absorption is usually unimportant, provided the total amount of drug absorbed is not markedly altered. On the other hand for drugs that are given as single doses, intended to be absorbed rapidly (e.g. hypnotics or analgesics), where a rapidly achieved high concentration is needed, a reduction in the rate of absorption may result in failure to achieve an adequate effect. ‘Table 1.1’, (p.2) lists some of the drug interactions that result from changes in absorption.
(a) Effects of changes in gastrointestinal pH
The passage of drugs through mucous membranes by simple passive diffusion depends upon the extent to which they exist in the non ionised lipid soluble form. Absorption is therefore governed by the pKa of the drug, its lipid solubility, the pH of the contents of the gut and various other parameters
relating to the pharmaceutical formulation of the drug. Thus the absorption of salicylic acid by the stomach is much greater at low pH than at high. On theoretical grounds it might be expected that alterations in gastric pH caused by drugs such as the H2-receptor antagonists would have a marked effect on absorption, but in practice the outcome is often uncertain because a number of other mechanisms may also come into play, such as chelation, adsorption and changes in gut motility, which can considerably affect what actually happens. However, in some cases the effect can be significant. Rises in pH due to ‘proton pump inhibitors’, (p.218), ‘H2-receptor antagonists’, (p.217) can markedly reduce the absorption of ketoconazole.
relating to the pharmaceutical formulation of the drug. Thus the absorption of salicylic acid by the stomach is much greater at low pH than at high. On theoretical grounds it might be expected that alterations in gastric pH caused by drugs such as the H2-receptor antagonists would have a marked effect on absorption, but in practice the outcome is often uncertain because a number of other mechanisms may also come into play, such as chelation, adsorption and changes in gut motility, which can considerably affect what actually happens. However, in some cases the effect can be significant. Rises in pH due to ‘proton pump inhibitors’, (p.218), ‘H2-receptor antagonists’, (p.217) can markedly reduce the absorption of ketoconazole.
(b) Adsorption, chelation and other complexing mechanisms
Activated charcoal is intended to act as an adsorbing agent within the gut for the treatment of drug overdose or to remove other toxic materials, but inevitably it can affect the absorption of drugs given in therapeutic doses. Antacids can also adsorb a large number of drugs, but often other mechanisms of interaction are also involved. For example, the tetracycline antibacterials can chelate with a number of divalent and trivalent metallic ions, such as calcium, aluminium, bismuth and iron, to form complexes that are both poorly absorbed and have reduced antibacterial effects (see ‘Figure 1.1’, (below)).
These metallic ions are found in dairy products and antacids. Separating the dosages by 2 to 3 hours goes some way towards reducing the effects of this type of interaction. The marked reduction in the bioavailability of penicillamine caused by some antacids seems also to be due to chelation, although adsorption may have some part to play. Colestyramine, an anionic exchange resin intended to bind bile acids and cholesterol metabolites in the gut, binds to a considerable number of drugs (e.g. digoxin, warfarin, levothyroxine), thereby reducing their absorption. ‘Table 1.1’, (p.2) lists some drugs that chelate, complex or adsorb other drugs.
(c) Changes in gastrointestinal motility
Since most drugs are largely absorbed in the upper part of the small intestine, drugs that alter the rate at which the stomach empties can affect absorption. Propantheline, for example, delays gastric emptying and reduces ‘paracetamol (acetaminophen)’ absorption, (p.192), whereas ‘metoclopramide’, (p.191), has the opposite effect. However, the total amount of drug absorbed remains unaltered. Propantheline also increases the absorption of ‘hydrochlorothiazide’, (p.959). Drugs with antimuscarinic effects decrease the motility of the gut, thus the tricyclic antidepressants can increase the absorption of ‘dicoumarol’, (p.457), probably because they increase the time available for dissolution and absorption but in the case of ‘levodopa’, (p.690), they may reduce the absorption, possibly because the exposure time to intestinal mucosal metabolism is increased. The same reduced levodopa absorption has also been seen with ‘homatropine’, (p.682). These examples illustrate that what actually happens is sometimes very unpredictable because the final outcome may be the result of several different mechanisms.
(d) Induction or inhibition of drug transporter proteins
The oral bioavailability of some drugs is limited by the action of drug transporter proteins, which eject drugs that have diffused across the gut lining back into the gut. At present, the most well characterised drug transporter is ‘P-glycoprotein’, (p.8). Digoxin is a substrate of P-glycoprotein, and drugs that induce this protein, such as rifampicin, may reduce the bioavailability of ‘digoxin’, (p.938).
(e) Malabsorption caused by drugs
Neomycin causes a malabsorption syndrome, similar to that seen with non-tropical sprue. The effect is to impair the absorption of a number of drugs including ‘digoxin’, (p.906) and ‘methotrexate’, (p.642).
1.2. Drug distribution interactions
(a) Protein-binding interactions
Following absorption, drugs are rapidly distributed around the body by the circulation. Some drugs are totally dissolved in the plasma water, but many others are transported with some proportion of their molecules in solution and the rest bound to plasma proteins, particularly the albumins.
The extent of this binding varies enormously but some drugs are extremely highly bound. For example, dicoumarol has only four out of every 1000 molecules remaining unbound at serum concentrations of 0.5 mg%. Drugs can also become bound to albumin in the interstitial fluid, and some, such as digoxin, can bind to the heart muscle tissue.
The binding of drugs to the plasma proteins is reversible, an equilibrium being established between those molecules that are bound and those that are not. Only the unbound molecules remain free and pharmacologically active, while those that are bound form a circulating but pharmacologically inactive reservoir which, in the case of drugs with a low-extraction ratio, is temporarily protected from metabolism and excretion. As the free molecules become metabolised, some of the bound molecules become unbound and pass into solution to exert their normal pharmacological actions, before they, in their turn are metabolised and excreted.
The extent of this binding varies enormously but some drugs are extremely highly bound. For example, dicoumarol has only four out of every 1000 molecules remaining unbound at serum concentrations of 0.5 mg%. Drugs can also become bound to albumin in the interstitial fluid, and some, such as digoxin, can bind to the heart muscle tissue.
The binding of drugs to the plasma proteins is reversible, an equilibrium being established between those molecules that are bound and those that are not. Only the unbound molecules remain free and pharmacologically active, while those that are bound form a circulating but pharmacologically inactive reservoir which, in the case of drugs with a low-extraction ratio, is temporarily protected from metabolism and excretion. As the free molecules become metabolised, some of the bound molecules become unbound and pass into solution to exert their normal pharmacological actions, before they, in their turn are metabolised and excreted.

Fig. 1.1 A drug chelation interaction. Tetracycline forms a less-soluble chelate with iron if the two drugs are allowed to mix within the gut. This reduces the absorption and depresses the serum levels and the antibacterial effects (after Neuvonen PJ, BMJ (1970) 4, 532, with permission). The same interaction can occur with other ions such as Al3+, Ca2+, Mg2+, Bi2+ and Zn2+.
Depending on the concentrations and their relative affinities for the binding sites, one drug may successfully compete with another and displace it from the sites it is already occupying. The displaced (and now active) drug molecules pass into the plasma water where their concentration rises. So for example, a drug that reduces the binding from 99 to 95% would increase the unbound concentration of free and active drug from 1 to 5% (a fivefold increase). This displacement is only likely to raise the number of free and active molecules significantly if the majority of the drug is within the plasma rather than the tissues, so that only drugs with a low apparent volume of distribution (Vd) will be affected. Examples include the sulphonylureas, such as tolbutamide (96% bound, Vd 10 litres), oral anticoagulants, such as warfarin (99% bound, Vd 9 litres), and phenytoin (90% bound, Vd 35 litres). However, another important factor is clearance. Clinically important protein-binding interactions are unlikely if only a small proportion of the drug is eliminated during a single-passage through the eliminating organ (low-extraction ratio drugs), since any increase in free fraction will be effectively cleared. Most drugs that are extensively bound to plasma proteins and subject to displacement reactions (e.g. warfarin, sulphonylureas, phenytoin, methotrexate, and valproate) have lowextraction ratios, and drug exposure is therefore independent of proteinbinding.
An example of displacement of this kind happens when patients stabilised on warfarin are given cloral hydrate because its major metabolite, trichloroacetic acid, is a highly bound compound that successfully displaces warfarin. This effect is only very short-lived because the now free and active warfarin molecules become exposed to metabolism as the blood flows through the liver, and the amount of drug rapidly falls. This transient increase in free warfarin levels is unlikely to change the anticoagulant effect of warfarin because the clotting factor complexes that are produced when warfarin is taken have a very long half-life, and thus take a long time to reach a new steady state. Normally no change in the warfarin dosage is needed (see ‘Coumarins + Cloral and derivatives’, p.396).
In vitro many commonly used drugs are capable of being displaced by others but in the body the effects seem almost always to be buffered so effectively that the outcome is not normally clinically important. It would therefore seem that the importance of this interaction mechanism has been grossly over-emphasised,1-3 It is difficult to find an example of a clinically important interaction due to this mechanism alone. It has been suggested that this interaction mechanism is likely to be important only for drugs given intravenously that have a high-extraction ratio, a short pharmacokinetic-pharmacodynamic half-life and a narrow therapeutic index. Lidocaine has been given as an example of a drug fitting these criteria.3 Some drug interactions that were originally assumed to be due to changes in protein binding have subsequently been shown to have other interaction mechanisms involved. For example, inhibition of metabolism has subsequently been shown to be important in the interactions between ‘warfarin and phenylbutazone’, (p.434), and ‘tolbutamide and sulphonamide’, (p.506).
However, knowledge of altered protein binding is important in therapeutic drug monitoring. Suppose for example a patient taking phenytoin was given a drug that displaced phenytoin from its binding sites. The amount of free phenytoin would rise but this would be quickly eliminated by metabolism
and excretion thereby keeping the amount of free active phenytoin the same. However, the total amount of phenytoin would now be reduced. Therefore if phenytoin was monitored using an assay looking at total phenytoin levels it may appear that the phenytoin is subtherapeutic and that the dose may therefore need increasing. However, as the amount of free active phenytoin is unchanged this would not be necessary and may even be dangerous.
Basic drugs as well as acidic drugs can be highly protein bound, but clinically important displacement interactions do not seem to have been described. The reasons seem to be that the binding sites within the plasma are different from those occupied by acidic drugs (alpha-1-acid glycoprotein rather than albumin) and, in addition, basic drugs have a large Vd with only a small proportion of the total amount of drug being within the plasma.
An example of displacement of this kind happens when patients stabilised on warfarin are given cloral hydrate because its major metabolite, trichloroacetic acid, is a highly bound compound that successfully displaces warfarin. This effect is only very short-lived because the now free and active warfarin molecules become exposed to metabolism as the blood flows through the liver, and the amount of drug rapidly falls. This transient increase in free warfarin levels is unlikely to change the anticoagulant effect of warfarin because the clotting factor complexes that are produced when warfarin is taken have a very long half-life, and thus take a long time to reach a new steady state. Normally no change in the warfarin dosage is needed (see ‘Coumarins + Cloral and derivatives’, p.396).
In vitro many commonly used drugs are capable of being displaced by others but in the body the effects seem almost always to be buffered so effectively that the outcome is not normally clinically important. It would therefore seem that the importance of this interaction mechanism has been grossly over-emphasised,1-3 It is difficult to find an example of a clinically important interaction due to this mechanism alone. It has been suggested that this interaction mechanism is likely to be important only for drugs given intravenously that have a high-extraction ratio, a short pharmacokinetic-pharmacodynamic half-life and a narrow therapeutic index. Lidocaine has been given as an example of a drug fitting these criteria.3 Some drug interactions that were originally assumed to be due to changes in protein binding have subsequently been shown to have other interaction mechanisms involved. For example, inhibition of metabolism has subsequently been shown to be important in the interactions between ‘warfarin and phenylbutazone’, (p.434), and ‘tolbutamide and sulphonamide’, (p.506).
However, knowledge of altered protein binding is important in therapeutic drug monitoring. Suppose for example a patient taking phenytoin was given a drug that displaced phenytoin from its binding sites. The amount of free phenytoin would rise but this would be quickly eliminated by metabolism
and excretion thereby keeping the amount of free active phenytoin the same. However, the total amount of phenytoin would now be reduced. Therefore if phenytoin was monitored using an assay looking at total phenytoin levels it may appear that the phenytoin is subtherapeutic and that the dose may therefore need increasing. However, as the amount of free active phenytoin is unchanged this would not be necessary and may even be dangerous.
Basic drugs as well as acidic drugs can be highly protein bound, but clinically important displacement interactions do not seem to have been described. The reasons seem to be that the binding sites within the plasma are different from those occupied by acidic drugs (alpha-1-acid glycoprotein rather than albumin) and, in addition, basic drugs have a large Vd with only a small proportion of the total amount of drug being within the plasma.
(b) Induction or inhibition of drug transport proteins
It is increasingly being recognised that distribution of drugs into the brain, and some other organs such as the testes, is limited by the action of drug transporter proteins such as P-glycoprotein. These proteins actively transport drugs out of cells when they have passively diffused in. Drugs that are inhibitors of these transporters could therefore increase the uptake of drug substrates into the brain, which could either increase adverse CNS effects, or be beneficial. For more information see ‘Drug transporter proteins’, (p.8).
1. MacKichan JJ. Protein binding drug displacement interactions. Fact or fiction? Clin Pharmacokinet
(1989) 16, 65–73.
2. Sansom LN, Evans AM. What is the true clinical significance of plasma protein binding displacement interactions? Drug Safety (1995) 12, 227–33.
3. Benet LZ, Hoener B-A. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther (2002) 71, 115–121.

Considered the preferred in vivo substrates, see Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective. J Clin Pharmacol (2003) 43, 443–69.
1.3. Drug metabolism (biotransformation) interactions
Although a few drugs are cleared from the body simply by being excreted unchanged in the urine, most are chemically altered within the body to less lipid-soluble compounds, which are more easily excreted by the kidneys. If this were not so, many drugs would persist in the body and continue to exert their effects for a long time. This chemical change is called ‘metabolism’, ‘biotransformation’, ‘biochemical degradation’ or sometimes ‘detoxification’. Some drug metabolism goes on in the serum, the kidneys, the skin and the intestines, but the greatest proportion is carried out by enzymes that are found in the membranes of the endoplasmic reticulum of the liver cells. If liver is homogenised and then centrifuged, the reticulum breaks up into small sacs called microsomes which carry the enzymes, and it is for this reason that the metabolising enzymes of the liver are frequently referred to as the ‘liver microsomal enzymes’.
We metabolise drugs by two major types of reaction. The first, so-called phase I reactions (involving oxidation, reduction or hydrolysis), turn drugs into more polar compounds, while phase II reactions involve coupling drugs with some other substance (e.g. glucuronic acid, known as glucuronidation) to make usually inactive compounds.
The majority of phase I oxidation reactions are carried out by the haemcontaining enzyme cytochrome P450. Cytochrome P450 is not a single entity, but is in fact a very large family of related isoenzymes, about 30 of which have been found in human liver tissue. However, in practice, only a few specific subfamilies seem to be responsible for most (about 90%) of the metabolism of the commonly used drugs. The most important isoenzymes are: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. Other enzymes involved in phase I metabolism include monoamine oxidases and epoxide hydrolases.
Less is known about the enzymes responsible for phase II conjugation reactions. However, UDP-glucuronyltransferases (UGT), methyltransferases, and N-acetyltransferases (NAT) are examples.
Although metabolism is very important in the body removing drugs, it is increasingly recognised that drugs can be adsorbed, distributed, or eliminated by transporters, the most well understood at present being ‘P-glycoprotein’, (p.8).
(a) Changes in first-pass metabolism
(i) Changes in blood flow through the liver
After absorption in the intestine, the portal circulation takes drugs directly to the liver before they are distributed by the blood flow around the rest of the body. A number of highly lipid-soluble drugs undergo substantial biotransformation during this first-pass through the gut wall and liver and there is some evidence that some drugs can have a marked effect on the extent of first pass metabolism by altering the blood flow through the liver. However, there are few clinically relevant examples of this, and many can be explained by other mechanisms, usually altered hepatic metabolism (see (ii) below). One possible example is the increase in rate of absorption of dofetilide with ‘verapamil’, (p.256), which has resulted in an increased incidence of torsade de pointes.
Another is the increase in bioavailability of high-extraction beta blockers with ‘hydralazine’, (p.847), possibly caused by altered hepatic blood flow, or altered metabolism.
(ii) Inhibition or induction of first-pass metabolism
The gut wall contains metabolising enzymes, principally the cytochrome P450 isoenzymes. In addition to the altered metabolism caused by changes in hepatic blood flow (see (i) above) there is evidence that some drugs can have a marked effect on the extent of first-pass metabolism by inhibiting or inducing the cytochrome P450 isoenzymes in the gut wall or in the liver.
An example is the effect of grapefruit juice, which seems to inhibit the cytochrome P450 isoenzyme CYP3A4, mainly in the gut, and therefore reduces the metabolism of oral calcium-channel blockers. Although altering the amount of drug ‘absorbed’, these interactions are usually considered drug metabolism interactions. The effect of grapefruit on the metabolism of other drugs is discussed further under ‘Drug-food interactions’, (p.11).
(b) Enzyme induction
When barbiturates were widely used as hypnotics it was found necessary to keep increasing the dosage as time went by to achieve the same hypnotic effect, the reason being that the barbiturates increase the activity of the microsomal enzymes so that extent of metabolism and excretion increases. This phenomenon of enzyme stimulation or ‘induction’ not only accounts for the need for an increased barbiturate dose but if another drug that is metabolised by the same range of enzymes is also present, its enzymatic metabolism is similarly increased and larger doses are needed to maintain the same therapeutic effect. However, note that not all enzyme-inducing drugs induce their own metabolism (a process known as auto-induction). The metabolic pathway that is most commonly induced is phase I oxidation mediated by the cytochrome P450 isoenzymes. The main drugs responsible for induction of the most clinically important cytochrome P450 isoenzymes are listed in ‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6). ‘Figure 1.2’, (see below) shows the reduction in trough ciclosporin levels when it is given with the enzyme inducer, St John’s wort. ‘St John’swort’, (p.1037), induces the metabolism of ciclosporin by induction of CYP3A4 and possibly also P-glycoprotein. ‘Figure 1.3’, (see above) shows the effects of another enzyme inducer, rifampicin (rifampin) on the serum levels of ‘ciclosporin’, (p.1022), presumably via its effects on CYP3A4. Phase II glucuronidation can also be induced. An example is when rifampicin induces the glucuronidation of ‘zidovudine’, (p.792).
The extent of the enzyme induction depends on the drug and its dosage, but it may take days or even 2 to 3 weeks to develop fully, and may persist for a similar length of time when the enzyme inducer is stopped. This means that enzyme induction interactions are delayed in onset and slow to resolve. Enzyme induction is a common mechanism of interaction and is not confined to drugs; it is also caused by the chlorinated hydrocarbon insecticides such as dicophane and lindane, and smoking tobacco.
If one drug reduces the effects of another by enzyme induction, it may be possible to accommodate the interaction simply by raising the dosage of the drug affected, but this requires good monitoring, and there are obvious hazards if the inducing drug is eventually stopped without remembering to reduce the dosage again. The raised drug dosage may be an overdose when the drug metabolism has returned to normal.
(c) Enzyme inhibition
More common than enzyme induction is the inhibition of enzymes. This results in the reduced metabolism of an affected drug, so that it may begin to accumulate within the body, the effect usually being essentially the same as when the dosage is increased. Unlike enzyme induction, which may take several days or even weeks to develop fully, enzyme inhibition can occur within 2 to 3 days, resulting in the rapid development of toxicity. The metabolic pathway that is most commonly inhibited is phase I oxidation by the cytochrome P450 isoenzymes. The main drugs responsible for inhibition of the most clinically important cytochrome P450 isoenzymes are listed in ‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6). For example a marked increase occurred in the plasma levels of a single dose of sildenafil after ritonavir had also been taken for 7 days, probably because ritonavir inhibits the metabolism of sildenafil by CYP3A4 (see ‘Phosphodiesterase type-5 inhibitors + Protease inhibitors’, p.1273).
An example of inhibition of phase I hydrolytic metabolism, is the inhibition of epoxide hydrolase by valpromide, which increases the levels of ‘carbamazepine’, (p.537). Phase II conjugative metabolism can also be inhibited.
Examples are the inhibition of carbamazepine glucuronidation by ‘sodium valproate’, (p.537), and the inhibition of methyltransferase by aminosalicylates causing raised levels of ‘azathioprine’, (p.665).
The clinical significance of many enzyme inhibition interactions depends on the extent to which the serum levels of the drug rise. If the serum levels remain within the therapeutic range the interaction may not be clinically important.

Fig. 1.2 An enzyme induction interaction. Chronology of ciclosporin trough concentrations (——) in a patient self-medicating with St John’s wort. ----------- = desired ciclosporin therapeutic range (after Barone GW, Gurley BJ, Ketel BL, Lightfoot ML, Abul-Ezz SR. Drug interaction between St. John’s Wort and Cyclosporine. Ann Pharmacother (2000) 34: 1013–16, with permission).

We metabolise drugs by two major types of reaction. The first, so-called phase I reactions (involving oxidation, reduction or hydrolysis), turn drugs into more polar compounds, while phase II reactions involve coupling drugs with some other substance (e.g. glucuronic acid, known as glucuronidation) to make usually inactive compounds.
The majority of phase I oxidation reactions are carried out by the haemcontaining enzyme cytochrome P450. Cytochrome P450 is not a single entity, but is in fact a very large family of related isoenzymes, about 30 of which have been found in human liver tissue. However, in practice, only a few specific subfamilies seem to be responsible for most (about 90%) of the metabolism of the commonly used drugs. The most important isoenzymes are: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. Other enzymes involved in phase I metabolism include monoamine oxidases and epoxide hydrolases.
Less is known about the enzymes responsible for phase II conjugation reactions. However, UDP-glucuronyltransferases (UGT), methyltransferases, and N-acetyltransferases (NAT) are examples.
Although metabolism is very important in the body removing drugs, it is increasingly recognised that drugs can be adsorbed, distributed, or eliminated by transporters, the most well understood at present being ‘P-glycoprotein’, (p.8).
(a) Changes in first-pass metabolism
(i) Changes in blood flow through the liver
After absorption in the intestine, the portal circulation takes drugs directly to the liver before they are distributed by the blood flow around the rest of the body. A number of highly lipid-soluble drugs undergo substantial biotransformation during this first-pass through the gut wall and liver and there is some evidence that some drugs can have a marked effect on the extent of first pass metabolism by altering the blood flow through the liver. However, there are few clinically relevant examples of this, and many can be explained by other mechanisms, usually altered hepatic metabolism (see (ii) below). One possible example is the increase in rate of absorption of dofetilide with ‘verapamil’, (p.256), which has resulted in an increased incidence of torsade de pointes.
Another is the increase in bioavailability of high-extraction beta blockers with ‘hydralazine’, (p.847), possibly caused by altered hepatic blood flow, or altered metabolism.
(ii) Inhibition or induction of first-pass metabolism
The gut wall contains metabolising enzymes, principally the cytochrome P450 isoenzymes. In addition to the altered metabolism caused by changes in hepatic blood flow (see (i) above) there is evidence that some drugs can have a marked effect on the extent of first-pass metabolism by inhibiting or inducing the cytochrome P450 isoenzymes in the gut wall or in the liver.
An example is the effect of grapefruit juice, which seems to inhibit the cytochrome P450 isoenzyme CYP3A4, mainly in the gut, and therefore reduces the metabolism of oral calcium-channel blockers. Although altering the amount of drug ‘absorbed’, these interactions are usually considered drug metabolism interactions. The effect of grapefruit on the metabolism of other drugs is discussed further under ‘Drug-food interactions’, (p.11).
(b) Enzyme induction
When barbiturates were widely used as hypnotics it was found necessary to keep increasing the dosage as time went by to achieve the same hypnotic effect, the reason being that the barbiturates increase the activity of the microsomal enzymes so that extent of metabolism and excretion increases. This phenomenon of enzyme stimulation or ‘induction’ not only accounts for the need for an increased barbiturate dose but if another drug that is metabolised by the same range of enzymes is also present, its enzymatic metabolism is similarly increased and larger doses are needed to maintain the same therapeutic effect. However, note that not all enzyme-inducing drugs induce their own metabolism (a process known as auto-induction). The metabolic pathway that is most commonly induced is phase I oxidation mediated by the cytochrome P450 isoenzymes. The main drugs responsible for induction of the most clinically important cytochrome P450 isoenzymes are listed in ‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6). ‘Figure 1.2’, (see below) shows the reduction in trough ciclosporin levels when it is given with the enzyme inducer, St John’s wort. ‘St John’swort’, (p.1037), induces the metabolism of ciclosporin by induction of CYP3A4 and possibly also P-glycoprotein. ‘Figure 1.3’, (see above) shows the effects of another enzyme inducer, rifampicin (rifampin) on the serum levels of ‘ciclosporin’, (p.1022), presumably via its effects on CYP3A4. Phase II glucuronidation can also be induced. An example is when rifampicin induces the glucuronidation of ‘zidovudine’, (p.792).
The extent of the enzyme induction depends on the drug and its dosage, but it may take days or even 2 to 3 weeks to develop fully, and may persist for a similar length of time when the enzyme inducer is stopped. This means that enzyme induction interactions are delayed in onset and slow to resolve. Enzyme induction is a common mechanism of interaction and is not confined to drugs; it is also caused by the chlorinated hydrocarbon insecticides such as dicophane and lindane, and smoking tobacco.
If one drug reduces the effects of another by enzyme induction, it may be possible to accommodate the interaction simply by raising the dosage of the drug affected, but this requires good monitoring, and there are obvious hazards if the inducing drug is eventually stopped without remembering to reduce the dosage again. The raised drug dosage may be an overdose when the drug metabolism has returned to normal.
(c) Enzyme inhibition
More common than enzyme induction is the inhibition of enzymes. This results in the reduced metabolism of an affected drug, so that it may begin to accumulate within the body, the effect usually being essentially the same as when the dosage is increased. Unlike enzyme induction, which may take several days or even weeks to develop fully, enzyme inhibition can occur within 2 to 3 days, resulting in the rapid development of toxicity. The metabolic pathway that is most commonly inhibited is phase I oxidation by the cytochrome P450 isoenzymes. The main drugs responsible for inhibition of the most clinically important cytochrome P450 isoenzymes are listed in ‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6). For example a marked increase occurred in the plasma levels of a single dose of sildenafil after ritonavir had also been taken for 7 days, probably because ritonavir inhibits the metabolism of sildenafil by CYP3A4 (see ‘Phosphodiesterase type-5 inhibitors + Protease inhibitors’, p.1273).
An example of inhibition of phase I hydrolytic metabolism, is the inhibition of epoxide hydrolase by valpromide, which increases the levels of ‘carbamazepine’, (p.537). Phase II conjugative metabolism can also be inhibited.
Examples are the inhibition of carbamazepine glucuronidation by ‘sodium valproate’, (p.537), and the inhibition of methyltransferase by aminosalicylates causing raised levels of ‘azathioprine’, (p.665).
The clinical significance of many enzyme inhibition interactions depends on the extent to which the serum levels of the drug rise. If the serum levels remain within the therapeutic range the interaction may not be clinically important.

Fig. 1.2 An enzyme induction interaction. Chronology of ciclosporin trough concentrations (——) in a patient self-medicating with St John’s wort. ----------- = desired ciclosporin therapeutic range (after Barone GW, Gurley BJ, Ketel BL, Lightfoot ML, Abul-Ezz SR. Drug interaction between St. John’s Wort and Cyclosporine. Ann Pharmacother (2000) 34: 1013–16, with permission).

Fig. 1.3 An enzyme induction interaction. Rifampicin (600 mg daily plus isoniazid) increased the metabolism of ciclosporin in this patient, thereby reducing the trough serum levels. He subsequently died because his heart transplant was rejected (after Transplant Proc, 16, Van Buren D, Wideman CA, Ried M, Gibbons S, Van Buren CT, Jarowenko M, Flechner SM, Frazier OH, Cooley DA, Kahan BD. The antagonistic effect of rifampicin upon cyclosporine bioavailability. 1642–5, Copyright Elsevier (1984)).


 *Considered the preferred in vivo substrates, see Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective.
*Considered the preferred in vivo substrates, see Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective.
J Clin Pharmacol (2003) 43, 443–69.


 *Considered the preferred in vivo substrates, see Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective. J Clin Pharmacol (2003) 43, 443–69.
*Considered the preferred in vivo substrates, see Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective. J Clin Pharmacol (2003) 43, 443–69.
(d) Genetic factors in drug metabolism
An increased understanding of genetics has shown that some of the cytochrome P450 isoenzymes are subject to ‘genetic polymorphism’, which simply means that some of the population have a variant of the isoenzyme with different (usually poor) activity. The best known example is CYP2D6, for which a small proportion of the population have the variant with low activity and are described as being poor or slow metabolisers (about 5 to 10% in white Caucasians, 0 to 2% in Asians and black people).
Which group any particular individual falls into is genetically determined.
The majority who possess the isoenzyme are called ‘fast or extensive metabolisers’. It is possible to find out which group any particular individual falls into by looking at the way a single dose of a test or ‘probe’ drug is metabolised. This varying ability to metabolise certain drugs may explain why some patients develop toxicity when given an interacting drug while others remain symptom free. CYP2D6, CYP2C9 and CYP2C19 also show polymorphism, whereas CYP3A4 does not, although there is still some broad variation in the population without there being distinct groups. The effects of CYP2C19 polymorphism are discussed in more detail in ‘Gastrointestinal drugs’, (p.960). At present, genotyping of cytochrome P450 isoenzymes is primarily a research tool and is not used clinically. In the future, it may become standard clinical practice and may be used to individualise drug therapy.1
‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6) are lists of drugs that are inhibitors, inducers, or substrates of the clinically important cytochrome P450 isoenzymes, and each drug has a cross reference to a monograph describing a drug interaction thought to occur via that mechanism.
If a new drug is shown to be an inducer, or an inhibitor, and/or a substrate of a given isoenzyme, these tables could be used to predict likely drug interactions. However, what may happen in vitro may not necessarily work in clinical practice because all of the many variables which can come into play are not known (such as how much of the enzyme is available, the concentration of the drug at the site of metabolism, and the affinity of the drug for the enzyme). Remember too that some drugs can be metabolised by more than one cytochrome P450 isoenzyme (meaning that this other isoenzyme may be able to ‘pick up’ more metabolism to compensate for the inhibited pathway); some drugs (and their metabolites) can both induce a particular isoenzyme and be metabolised by it; and some drugs (or their metabolites) can inhibit a particular isoenzyme but not be metabolised by it. With so many factors possibly impinging on the outcome of giving two or more drugs together, it is very easy to lose sight of one of the factors (or not even know about it) so that the sum of 2 plus 2 may not turn out to be the 4 that you have predicted.
For example, ritonavir and other protease inhibitors are well known potent inhibitors of CYP3A4, and in clinical use increase the levels of many drugs that are substrates of this isoenzyme. Methadone is a substrate of CYP3A4, and some in vitro data show that ritonavir (predictably) increased methadone levels. However, unexpectedly, in clinical use the protease inhibitors seem to decrease methadone levels, by a yet unknown mechanism (see, ‘Opioids; Methadone + Protease inhibitors’, p.182).
Another factor complicating the understanding of metabolic drug interactions is the finding that there is a large overlap between the inhibitors/inducers and substrates of P-glycoprotein (a ‘drug transporter protein’, (p.8)) and those of CYP3A4. Therefore, both mechanisms may be involved in many of the drug interactions previously thought to be due to effects on CYP3A4.
1. Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA (2001) 286, 2270–79.
2. Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a PhRMA perspective. J Clin Pharmacol (2003) 43, 443–69.
3. Bachmann KA, Ghosh R. The use of in vitro methods to predict in vivo pharmacokinetics and drug interactions. Curr Drug Metab (2001) 2, 299–314.
4. Yao C, Levy RH. Inhibition-based metabolic drug–drug interactions: predictions from in vitro data. J Pharm Sci (2002) 91, 1923–35.
5. Worboys PD, Carlile DJ. Implications and consequences of enzyme induction on preclinical and clinical drug development. Xenobiotica (2001) 31, 539–56.
6. Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Drug metabolism and drug interactions: application and clinical value of in vitro models. Curr Drug Metab (2003) 4, 423–59.
1.4. Drug excretion interactions
With the exception of the inhalation anaesthetics, most drugs are excreted either in the bile or in the urine. Blood entering the kidneys along the renal arteries is, first of all, delivered to the glomeruli of the tubules where molecules small enough to pass through the pores of the glomerular membrane (e.g. water, salts, some drugs) are filtered through into the lumen of the tubules.
Larger molecules, such as plasma proteins, and blood cells are retained within the blood. The blood flow then passes to the remaining parts of the kidney tubules where active energy-using transport systems are able to remove drugs and their metabolites from the blood and secrete them into the tubular filtrate. The renal tubular cells additionally possess active and passive transport systems for the reabsorption of drugs. Interference by drugs with renal tubular fluid pH, with active transport systems and with blood flow to the kidney can alter the excretion of other drugs.
 Fig. 1.4 An excretion interaction. If the tubular filtrate is acidified, most of the molecules of weakly acid drugs (HX) exist in an un-ionised lipid-soluble form and are able to return through the lipid membranes of the tubule cells by simple diffusion. Thus they are retained. In alkaline urine most of the drug molecules exist in an ionised non-lipid soluble form (X). In this form the molecules are unable to diffuse freely through these membranes and are therefore lost in the urine.
Fig. 1.4 An excretion interaction. If the tubular filtrate is acidified, most of the molecules of weakly acid drugs (HX) exist in an un-ionised lipid-soluble form and are able to return through the lipid membranes of the tubule cells by simple diffusion. Thus they are retained. In alkaline urine most of the drug molecules exist in an ionised non-lipid soluble form (X). In this form the molecules are unable to diffuse freely through these membranes and are therefore lost in the urine.
(a) Changes in urinary pH
As with drug absorption in the gut, passive reabsorption of drugs depends upon the extent to which the drug exists in the non-ionised lipid-soluble form, which in its turn depends on its pKa and the pH of the urine. Only the non-ionised form is lipid-soluble and able to diffuse back through the lipid membranes of the tubule cells. Thus at high pH values (alkaline), weakly acid drugs (pKa 3 to 7.5) largely exist as ionised lipid-insoluble molecules, which are unable to diffuse into the tubule cells and will there fore remain in the urine and be removed from the body. The converse will be true for weak bases with pKa values of 7.5 to 10.5. Thus pH changes that increase the amount in the ionised form (alkaline urine for acidic drugs, acid urine for basic drugs) increase the loss of the drug, whereas moving the pH in the opposite direction will increase their retention. ‘Figure 1.4’, (p.7) illustrates the situation with a weakly acidic drug. The clinical significance of this interaction mechanism is small, because although a very large number of drugs are either weak acids or bases, almost all are largely metabolised by the liver to inactive compounds and few are excreted in the urine unchanged. In practice therefore only a handful of drugs seem to be affected by changes in urinary pH (possible exceptions include changes in the excretion of ‘quinidine’, (p.277) or ‘analgesic-dose aspirin’, (p.135), due to alterations in urinary pH caused by antacids, and the increase in the clearance of ‘methotrexate’, (p.654), with urinary alkalinisers). In cases of overdose, deliberate manipulation of urinary pH has been used to increase the removal of drugs such as methotrexate and salicylates.

(b) Changes in active renal tubular excretion
Drugs that use the same active transport systems in the renal tubules can compete with one another for excretion. For example, probenecid reduces the excretion of penicillin and other drugs. With the increasing understanding of drug transporter proteins in the kidneys, it is now known that probenecid inhibits the renal secretion of many other anionic drugs by organic anion transporters (OATs).1 Probenecid possibly also inhibits some of the ABC transporters in the kidneys. The ABC transporter, P-glycoprotein, is also present in the kidneys, and drugs that alter this may alter renal drug elimination. See, ‘Drug transporter proteins’, (p.8), for further discussion. Some examples of drugs that possibly interact by alterations in renal transport are given in ‘Table 1.5’, (see above).
(c) Changes in renal blood flow
The flow of blood through the kidney is partially controlled by the production of renal vasodilatory prostaglandins. If the synthesis of these prostaglandins is inhibited the renal excretion of some drugs may be reduced. An interaction where this is the suggested mechanism is the rise in serum lithium seen with some NSAIDs, see ‘Lithium + NSAIDs’, p.1125.
(d) Biliary excretion and the entero-hepatic shunt
(i) Enterohepatic recirculation
A number of drugs are excreted in the bile, either unchanged or conjugated (e.g. as the glucuronide) to make them more water soluble. Some of the conjugates are metabolised to the parent compound by the gut flora and are then reabsorbed. This recycling process prolongs the stay of the drug within the body, but if the gut flora are diminished by the presence of an antibacterial, the drug is not recycled and is lost more quickly. This may possibly explain the rare failure of the oral contraceptives that can be brought about by the concurrent use of penicillins or tetracyclines, but see
Mechanism in ‘Hormonal contraceptives + Antibacterials; Penicillins’, p.981. Antimicrobial-induced reductions in gut bacteria may reduce the activation of ‘sulfasalazine’, (p.973).
(ii) Drug transporters
Increasing research shows that numerous drug transporter proteins (both from the ABC family and SLC family, see ‘Drug transporter proteins’, (see below)) are involved in the hepatic extraction and secretion of drugs into the bile.2 The relevance of many of these to drug interactions is still unclear, but the bile salt export pump (ABCB11) is known to be inhibited by a variety of drugs including ciclosporin, glibenclamide, and bosentan.
Inhibition of this pump may increase the risk of cholestasis, and the manufacturer of bosentan says that they should be avoided in patients taking bosentan (see ‘glibenclamide’, (p.515) and ‘ciclosporin’, (p.1026)).
1. Lee W, Kim RB. Transporters and renal drug elimination. Annu Rev Pharmacol Toxicol (2004) 44, 137–66.
2. Faber KN, Müller M, Jansen PLM. Drug transport proteins in the liver. Adv Drug Deliv Rev (2003) 55, 107–24.
1.5. Drug transporter proteins
Drugs and endogenous substances are known to cross biological membranes, not just by passive diffusion, but by carrier-mediated processes, often known as transporters. Significant advances in the identification of various transporters have been made, although the contribution of many of these to drug interactions in particular, is still unclear.1,2 The most well known is P-glycoprotein, which is a product of the MDR1 gene (ABCB1 gene) and a member of the ATP-binding cassette (ABC) family of efflux transporters.1 Its involvement in drug interactions is discussed in (a) below.
Another ABC transporter is sister P-glycoprotein, otherwise called the bile salt export pump (BSEP or ABCB11).1 It has been suggested that inhibition of this pump may increase the risk of cholestasis, see Drug transporters under ‘Drug excretion interactions’, (p.7).
Other transporters that are involved in some drug interactions are the organic anion transporters (OATs), organic anion-transporting polypeptides (OATPs) and organic cation transporters (OCTs), which are members of the solute carrier superfamily (SLC) of transporters.1 The best known example
of an OAT inhibitor is probenecid, which affects the renal excretion of a number of drugs, see Changes in active kidney tubule excretion under ‘Drug excretion interactions’, (p.7).
 1. Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol Rev (2003) 55, 425-61.
1. Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol Rev (2003) 55, 425-61.
(a) P-glycoprotein interactions
More and more evidence is accumulating to show that some drug interactions occur because they interfere with the activity of P-glycoprotein. This is an efflux pump found in the membranes of certain cells, which can push metabolites and drugs out of the cells and have an impact on the extent of drug absorption (via the intestine), distribution (to the brain, testis, or placenta) and elimination (in the urine and bile). So, for example, the P-glycoprotein in the cells of the gut lining can eject some already-absorbed drug molecules back into the intestine resulting in a reduction in the total amount of drug absorbed. In this way P-glycoprotein acts as a barrier to absorption. The activity of P-glycoprotein in the endothelial cells of the blood-brain barrier can also eject certain drugs from the brain, limiting CNS penetration and effects.
The pumping actions of P-glycoprotein can be induced or inhibited by some drugs. So for example, the induction (or stimulation) of the activity of P-glycoprotein by rifampicin (rifampin) within the lining cells of the gut causes digoxin to be ejected into the gut more vigorously. This results in a fall in the plasma levels of digoxin (see ‘Digitalis glycosides + Rifamycins’, p.938). In contrast, verapamil appears to inhibit the activity of P-glycoprotein, and is well known to increase digoxin levels (see ‘Digitalis glycosides + Calcium-channel blockers; Verapamil’, p.916). Ketoconazole also has P-glycoprotein inhibitory effects, and has been shown to increase CSF levels of ritonavir, possibly by preventing the efflux of ritonavir from the CNS (see ‘Protease inhibitors + Azoles; Ketoconazole’, p.814). Thus the induction or inhibition of P-glycoprotein can have an impact on the pharmacokinetics of some drugs. Note that there is evidence that P-glycoprotein inhibition may have a greater impact on drug distribution (e.g. into the brain) than on drug absorption (e.g. plasma levels). 2 There is an overlap between CYP3A4 and P-glycoprotein inhibitors, inducers and substrates. Therefore, both mechanisms may be involved in many of the drug interactions traditionally thought to be due to changes in CYP3A4. ‘Table 1.6’, (p.8) lists some possible P-glycoprotein inhibitors and inducers. Many drugs that are substrates for CYP3A4 (see ‘Table 1.4’, (p.6)) are also substrates for P-glycoprotein. Digoxin and talinolol are examples of the few drugs that are substrates for P-glycoprotein but not CYP3A4.
P-glycoprotein is also expressed in some cancer cells (where it was first identified). This has led to the development of specific P-glycoprotein inhibitors, such as valspodar, with the aim of improving the penetration of cytotoxic drugs into cancer cells.
1. Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery
and development. Pharmacol Rev (2003) 55, 425–61.
2. Lin JH, Yamazaki M. Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev
(2003) 35, 417–54.
2. Pharmacodynamic interactions
Pharmacodynamic interactions are those where the effects of one drug are changed by the presence of another drug at its site of action. Sometimes the drugs directly compete for particular receptors (e.g. beta2 agonists, such as salbutamol, and beta blockers, such as propranolol) but often the reaction is more indirect and involves interference with physiological mechanisms. These interactions are much less easy to classify neatly than those of a pharmacokinetic type.
2.1. Additive or synergistic interactions
If two drugs that have the same pharmacological effect are given together the effects can be additive. For example, alcohol depresses the CNS and, if taken in moderate amounts with normal therapeutic doses of any of a large number of drugs (e.g. anxiolytics, hypnotics, etc.), may cause excessive drowsiness. Strictly speaking (as pointed out earlier) these are not interactions within the definition given in ‘What is a drug interaction?’, (p.1). Nevertheless, it is convenient to consider them within the broad context of the clinical outcome of giving two drugs together.
Additive effects can occur with both the main effects of the drugs as well as their adverse effects, thus an additive ‘interaction’ can occur with antimuscarinic antiparkinson drugs (main effect) or butyrophenones (adverse effect) that can result in serious antimuscarinic toxicity (see ‘Antipsychotics + Antimuscarinics’, p.708).
Sometimes the additive effects are solely toxic (e.g. additive ototoxicity, nephrotoxicity, bone marrow depression, QT interval prolongation). Examples of these reactions are listed in ‘Table 1.7’, (see below). It is common to use the terms ‘additive’, ‘summation’, ‘synergy’ or ‘potentiation’ to describe what happens if two or more drugs behave like this. These words have precise pharmacological definitions but they are often used rather loosely as synonyms because in practice it is often very difficult to know the extent of the increased activity, that is to say whether the effects are greater or smaller than the sum of the individual effects.
The serotonin syndrome
In the 1950s a serious and life-threatening toxic reaction was reported in patients taking iproniazid (an MAOI) when they were given ‘pethidine (meperidine)’, (p.1140). The reasons were then not understood and even now we do not have the full picture. What happened is thought to have been due to over-stimulation of the 5-HT1A and 5-HT2A receptors and possibly other serotonin receptors in the central nervous system (in the brain stem and spinal cord in particular) due to the combined effects of these two drugs. It can occur exceptionally after taking only one drug, which causes over-stimulation of these 5-HT receptors, but much more usually it develops when two or more drugs (so-called serotonergic or serotomimetic drugs) act in concert. The characteristic symptoms (now known as the serotonin syndrome) fall into three main areas, namely altered mental status (agitation, confusion, mania), autonomic dysfunction (diaphoresis, diarrhoea, fever, shivering) and neuromuscular abnormalities (hyperreflexia, incoordination, myoclonus, tremor). These are the ‘Sternbach diagnostic criteria’ named after Dr Harvey Sternbach who drew up this list of clinical features and who suggested that at least three of them need to be seen before classifying this toxic reaction as the serotonin syndrome rather than the neuroleptic malignant syndrome.1
The syndrome can develop shortly after one serotonergic drug is added to another, or even if one is replaced by another without allowing a long enough washout period in between, and the problem usually resolves within about 24 hours if both drugs are withdrawn and supportive measures given. Non-specific serotonin antagonists (cyproheptadine, chlorpromazine, methysergide) have also been used for treatment. Most patients recover uneventfully, but there have been a few fatalities.

Following the first report of this syndrome, many other cases have been described involving ‘tryptophan and MAOIs’, (p.1151), the ‘tricyclic antidepressants and MAOIs’, (p.1149), and, more recently, the ‘SSRIs’, (p.1142) but other serotonergic drugs have also been involved and the list
continues to grow.
It is still not at all clear why many patients can take two, or sometimes several serotonergic drugs together without problems, while a very small number develop this serious toxic reaction, but it certainly suggests that there are as yet other factors involved that have yet to be identified. The full story is likely to be much more complex than just the simple additive effects of two drugs.
1. Sternbach H. The serotonin syndrome. Am J Psychiatry (1991) 148, 705–13.
2.2. Antagonistic or opposing interactions
In contrast to additive interactions, there are some pairs of drugs with activities that are opposed to one another. For example the coumarins can prolong the blood clotting time by competitively inhibiting the effects of dietary vitamin K. If the intake of vitamin K is increased, the effects of the oral anticoagulant are opposed and the prothrombin time can return to normal, thereby cancelling out the therapeutic benefits of anticoagulant treatment (see ‘Coumarins and related drugs + Vitamin K substances’, p.458).
Other examples of this type of interaction are listed in ‘Table 1.8’, (see below).

2.3. Drug or neurotransmitter uptake interactions
A number of drugs with actions that occur at adrenergic neurones can be prevented from reaching those sites of action by the presence of other drugs. The tricyclic antidepressants prevent the reuptake of noradrenaline (norepinephrine) into peripheral adrenergic neurones. Thus patients taking tricyclics and given parenteral noradrenaline have a markedly increased response (hypertension, tachycardia); see ‘Tricyclic antidepressants + Inotropes and Vasopressors’, p.1237. Similarly, the uptake of guanethidine (and related drugs guanoclor, betanidine, debrisoquine, etc.) is blocked by ‘chlorpromazine, haloperidol, tiotixene’, (p.887), a number of ‘amfetamine-like drugs’, (p.886) and the ‘tricyclic antidepressants’, (p.888) so that the antihypertensive effect is prevented. The antihypertensive effects of clonidine are also prevented by the tricyclic antidepressants, one possible reason being that the uptake of clonidine within the CNS is blocked (see ‘Clonidine + Tricyclic and related antidepressants’, p.884). Some of these interactions at adrenergic neurones are illustrated in ‘Figure 1.5’, (see below).

Fig. 1.5 Interactions at adrenergic neurones. A highly simplified composite diagram of an adrenergic neurone (molecules of noradrenaline (norepinephrine) indicated as (•) contained in a single vesicle at the nerve-ending) to illustrate in outline some of the different sites where drugs can interact. More details of these interactions are to be found in individual monographs.
(''The MAOIs inactivate monoamine oxidase and cause the accumulation of noradrenaline at the nerve ending.
When released by indirectly-acting sympathomimetics this results in a massive stimulation of the receptors and a grossly exaggerated pressor response")
("The tricyclic antidepressants, chlorpromazine, haloperidol, tiotixene, mazindol (?) and pizotifen (?) prevent the uptake of guanethidine and related drugs into the neurones, thereby blocking the antihypertensive effects")
("The tricyclic antidepressants block the uptake mechanism by which noradrenaline is taken into the neurone and removed from the receptor area. As a result the effects of administered noradrenaline are exaggerated")
("Alpha and beta blockers occupy the receptors and prevent the normal stimulant activity of the noradrenaline. Alpha blockers such as phentolamine will block the pressor effects of noradrenaline. Propranolol and similar nonselective beta blockers will antagonise the bronchodilator effects of betaagonist bronchodilators (e.g. salbutamol) ")
("Indirectly-acting sympathomimetics stimulate the release of noradrenaline")
("Directly-acting sympathomimetics act like noradrenaline by direct stimulation of the receptors")
("Mixed action sympathomimetics have both direct and indirect activity")
("Adrenergic nerve ending")
E. Drug-herb interactions
The market for herbal medicines and supplements in the Western world has markedly increased in recent years, and, not surprisingly, reports of interactions with ‘conventional’ drugs have arisen. The most well known and documented example is the interaction of St John’s wort (Hypericum perforatum) with a variety of drugs, see below. There have also been isolated reports of other herbal drug interactions, attributable to various mechanisms, including additive pharmacological effects.
Based on these reports, there are a growing number of reviews of herbal medicine interactions, which seek to predict likely interactions based on the, often hypothesised, actions of various herbs. Many of these predictions seem tenuous at best.
Rather than add to the volume of predicted interactions, at present, Stockley’s Drug Interactions includes only those interactions for which there are published reports.
To aid collection of data in this area, health professionals should routinely ask patients about their use of herbal medicines and supplements, and report any unexpected responses to treatment.
An additional problem in interpreting these interactions, is that the interacting constituent of the herb is usually not known and is therefore not standardised for. It could vary widely between different products, and batches of the same product.
St John’s wort
An increasing number of reports have implicated St John’s wort (Hypericum perforatum) in drug interactions. Evidence has shown that the herb can induce the cytochrome P450 isoenzyme CYP3A4, and can also induce ‘P-glycoprotein’, (p.8). Hence St John’s wort decreases the levels of ‘ciclosporin’, (p.1037) and ‘digoxin’, (p.927), respectively. Other less certain evidence suggests that CYP2E1 and CYP1A2 may also be induced.
St John’s wort has serotonergic properties, and this has resulted in a pharmacodynamic interaction with the ‘SSRIs’, (p.1224), namely the development of the serotonin syndrome. St John’s wort contains many possible constituents that could be responsible for its pharmacological effects. The major active constituents are currently considered to be hyperforin (a phloroglucinol) and hypericin (a naphthodianthrone). Hypericin is the only constituent that is standardised for, and then only in some St John’s wort preparations.
General references
1. Miller LG. Herbal medicinals. Selected clinical considerations focusing on known or potential drug-herb interactions. Arch Intern Med (1998) 158, 2200–11.
2. Fugh-Berman A. Herb-drug interactions. Lancet (2000) 355, 134–8. Correction. ibid. 1020.
3. Wang Z, Gorski JC, Hamman MA, Huang S-M, Lesko LJ, Hall SD. The effects of St John’s wort (Hypericum perforatum) on human cytochrome P450 activity. Clin Pharmacol Ther (2001) 70, 317–26.
4. Williamson EM. Drug interactions between herbal and prescription medicines. Drug Safety (2003) 26, 1075–92.
5. Henderson L, Yue QY, Bergquist C, Gerden B, Arlett P. St John’s wort (Hypericum perforatum): drug interactions and clinical outcomes. Br J Clin Pharmacol (2002) 54, 349–56.
6. Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Cui Y, Ang CYW. Cytochrome P450 phenotypic ratios for predicting herb-drug interactions in humans. Clin Pharmacol Ther (2002) 72, 276–87.
7. Dresser GK, Schwarz UI, Wilkinson GR, Kim RB. Coordinate induction of both cytochrome P4503A and MDR1 by St John’s wort in healthy subjects. Clin Pharmacol Ther (2003) 73, 41–50
F. Drug-food interactionsa
It is well established that food can cause clinically important changes in drug absorption through effects on gastrointestinal motility or by drug binding, see ‘Drug absorption interactions’, (p.3). In addition, it is well known that tyramine (present in some foodstuffs) may reach toxic concentrations in patients taking ‘MAOIs’, (p.1153). With the growth in understanding of drug metabolism mechanisms, it has been increasingly recognised that some foods can alter drug metabolism. Currently, grapefruit juice causes the most clinically relevant of these interactions, see (b) below.
(a) Cruciferous vegetables and charcoal-broiled meats
Cruciferous vegetables, such as brussels sprouts, cabbage, and broccoli, contain substances that are inducers of the cytochrome P450 isoenzyme CYP1A2. Chemicals formed by ‘burning’ meats additionally have these properties. These foods do not appear to cause any clinically important drug interactions in their own right, but their consumption may add another variable to drug interaction studies, so complicating interpretation. In drug interaction studies where alteration of CYP1A2 is a predicted mechanism, it may be better for patients to avoid these foods during the study.
(b) Grapefruit juice
By chance, grapefruit juice was chosen to mask the taste of alcohol in a study of the effect of alcohol on felodipine, which led to the discovery that grapefruit juice itself markedly increased felodipine levels, see ‘Calciumchannel blockers + Grapefruit juice’, p.869. In general, grapefruit juice inhibits intestinal CYP3A4, and only slightly affects hepatic CYP3A4. This is demonstrated by the fact that intravenous preparations of drugs that are metabolised by CYP3A4 are not much affected, whereas oral preparations of the same drugs are. These interactions result in increased drug levels.
Some drugs that are not metabolised by CYP3A4 show decreased levels with grapefruit juice, such as ‘fexofenadine’, (p.588). The probable reason for this is that grapefruit juice is an inhibitor of some drug transporters (see ‘Drug transporter proteins’, (p.8)), and possibly affects organic aniontransporting polypeptides (OATPs), although inhibition of P-glycoprotein has also been suggested.
The active constituent of grapefruit juice is uncertain. Grapefruit contains naringin, which degrades during processing to naringenin, a substance known to inhibit CYP3A4. Because of this, it has been assumed that whole grapefruit will not interact, but that processed grapefruit juice will. However, subsequently some reports have implicated the whole fruit. Other possible active constituents in the whole fruit include bergamottin and dihydroxybergamottin.
General references
1. Ameer B, Wientraub RA. Drug interactions with grapefruit juice. Clin Pharmacokinet (1997) 33, 103–21.
G. Conclusions
It is now quite impossible to remember all the known clinically important interactions and how they occur, which is why this reference publication has been produced, but there are some broad general principles that need little memorising:
• Be on the alert with any drugs that have a narrow therapeutic window or where it is necessary to keep serum levels at or above a suitable level (e.g. anticoagulants, antidiabetic drugs, antiepileptics, antihypertensives, anti-infectives, antineoplastic cytotoxics, digitalis glycosides, immunosuppressants, etc.).
• Remember some of those drugs that are key enzyme inducers (e.g. phenytoin, barbiturates, rifampicin, etc) or enzyme inhibitors (e.g. azole antifungals, HIV-protease inhibitors, erythromycin, SSRIs).
• Think about the basic pharmacology of the drugs under consideration so that obvious problems (additive CNS depression for example) are not overlooked, and try to think what might happen if drugs that affect the same receptors are used together. And don’t forget that many drugs affect more than one type of receptor.
• Keep in mind that the elderly are at risk because of reduced liver and renal function on which drug clearance depends.
2
ACE inhibitors + Allopurinol
Three cases of Stevens-Johnson syndrome (one fatal) and two cases of hypersensitivity have been attributed to the use of captopril with allopurinol. Anaphylaxis and myocardial infarction occurred in one man taking enalapril when given allopurinol. The combination of ACE inhibitors and allopurinol may increase the risk of leucopenia and serious infection, especially in renal impairment.
Clinical evidence
An elderly man with hypertension, chronic renal failure, congestive heart failure and mild polyarthritis receiving multiple drug treatment, which included captopril 25 mg twice daily and diuretics, developed fatal Stevens-Johnson syndrome about 5 weeks after starting to take allopurinol 100 mg twice daily.1 The authors of the report noted that the manufacturer of captopril was aware of two other patients who developed the syndrome 3 to 5 weeks after allopurinol was started.1 Another report describes fever, arthralgia and myalgia in a diabetic man with chronic renal failure who was also given captopril and allopurinol. He improved when the captopril was withdrawn.2 Exfoliatory facial dermatitis occurred in a patient with renal failure who was taking captopril and allopurinol.3 A man taking enalapril had an acute anaphylactic reaction with severe coronary spasm, culminating in myocardial infarction, within 20 minutes of taking allopurinol 100 mg. He recovered and continued to take enalapril without allopurinol.4
The UK manufacturer of captopril also warns that neutropenia and agranulocytosis, resulting in serious infection, have occurred in patients taking captopril and other ACE inhibitors, and that concurrent treatment with allopurinol may be a complicating factor, especially in those with renal impairment.5 However, the US manufacturer notes that, while renal impairment and a relatively high dose of captopril markedly increases the risk of neutropenia, no association between allopurinol and captopril and neutropenia has appeared in US reports.6
No significant pharmacokinetic changes were seen in 12 healthy subjects given allopurinol and captopril alone and in combination.7
Mechanism
Not understood. It is uncertain whether these are interactions because allopurinol alone can cause severe hypersensitivity reactions, particularly in the presence of renal failure and in conjunction with diuretic use. Captopril can also induce a hypersensitivity reaction.
Importance and management
These interactions are not clearly established, and the reaction appears to be rare and unpredictable. All that can be constructively said is that patients taking both drugs should be very closely monitored for any signs of hypersensitivity (e.g. skin reactions) or low white cell count (sore throat, fever), especially if they have renal impairment. The UK manufacturer of captopril recommends that differential white blood cell counts should be performed before adding allopurinol, then every 2 weeks during the first 3 months of treatment, and periodically thereafter.5 Similar caution and advice is given by the UK manufacturers of several other ACE inhibitors.
For other possible interactions with ACE inhibitors that might result in an increased risk of leucopenia see also ‘ACE inhibitors + Azathioprine’, p.18 and ‘ACE inhibitors + Procainamide’, p.33.
1. Pennell DJ, Nunan TO, O’Doherty MJ, Croft DN. Fatal Stevens-Johnson syndrome in a patient on captopril and allopurinol. Lancet (1984) i, 463.
2. Samanta A, Burden AC. Fever, myalgia, and arthralgia in a patient on captopril and allopurinol. Lancet (1984) i, 679.
3. Beeley L, Daly M, Stewart P. Bulletin of the West Midlands Centre for Adverse Drug Reaction Reporting (1987) 24, 9.
4. Ahmad S. Allopurinol and enalapril. Drug induced anaphylactic coronary spasm and acute myocardial infarction. Chest (1995) 108, 586.
5. Capoten (Captopril). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Capoten (Captopril). Par Pharmaceutical, Inc. US Prescribing information, June 2003.
7. Duchin KL, McKinstry DN, Cohen AI, Migdalof BH. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin Pharmacokinet (1988) 14, 241–59.
The combined use of ACE inhibitors and angiotensin II receptor antagonists increases the risk of hypotension, renal impairment and hyperkalaemia in patients with heart failure.
Clinical evidence, mechanism, importance and management
Both ACE inhibitors and angiotensin II receptor antagonists can have adverse renal effects and can cause hyperkalaemia. These effects might be expected to be additive when they are used together. In one randomised clinical study in patients with heart failure taking ACE inhibitors, the addition of candesartan resulted in higher rates of withdrawals than placebo for renal impairment (increase in creatinine 7.8% versus 4.1%) and hyperkalaemia (3.4% versus 0.7%).1 In another double-blind study in patients with heart failure, the combination of valsartan and captopril resulted in a higher incidence of adverse events leading to a dose reduction or a discontinuation of study treatment than either drug alone. For hypotension, treatment was discontinued in 90 (1.9%) of patients in the combined group, 70 (1.4%) of patients in the valsartan group, and 41 (0.8%) of patients in the captopril group. For renal causes the corresponding figures were 61 (1.3%), 53 (1.1%) and 40 (0.8%) of patients, respectively, and for hyperkalaemia the figures were 12 (0.2%), 7 (0.1%) and 4 (0.1%) of patients, respectively.2
Monitor renal function and serum potassium carefully when combination therapy is used.
1. McMurray JJV, Östergren J, Swedberg K, Granger CB, Held P, Michelson EL, Olofsson B, Yusuf S, Pfeffer MA; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin- converting-enzyme inhibitors: the CHARM-Added trial. Lancet (2003) 362, 767–71.
2. Pfeffer MA, McMurray JJV, Velazquez EJ, Rouleau J-L, Køber L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med (2003) 349, 1893–1906.
ACE inhibitors + Antacids
An aluminium/magnesium hydroxide antacid reduced the bioavailability of captopril by 40%, but this did not seem to be clinically important. The bioavailability of fosinopril was reduced by about one-third by Mylanta. An antacid did not affect ramipril pharmacokinetics.
Clinical evidence
In 10 healthy subjects an antacid containing aluminium/magnesium hydroxide and magnesium carbonate reduced the AUC of a single 50-mg dose of captopril by about 40%, when compared with the fasting state.
However, this did not alter the extent of the reduction in blood pressure.1
Another study found that Mylanta [aluminium/magnesium hydroxide and simeticone2] reduced the bioavailability of fosinopril 20 mg by about one-third.3
It is briefly noted in a review that antacid use did not affect the pharmacokinetics of ramiprilat, the active metabolite of ramipril.4
Mechanism
The mechanism of this interaction is uncertain, but is unlikely to be due to elevated gastric pH since cimetidine did not have a similar effect.3
Importance and management
Note that greater decreases in captopril bioavailability (caused by ‘food’, (p.26)) were found not to be clinically relevant, therefore, it is unlikely the change seen with antacids will be clinically important.
However, with fosinopril, the manufacturers2,5 suggest separating administration of antacids by at least 2 hours.
The UK manufacturers of quinapril6 and trandolapril7 also warn that antacids may reduce the bioavailability of ACE inhibitors, quite possibly based on the way these named ACE inhibitors interact, but there seems to be no evidence of a clinically significant interaction in practice.
1. Mäntylä R, Männistö PT, Vuorela A, Sundberg S, Ottoila P. Impairment of captopril bioavailability by concomitant food and antacid intake. Int J Clin Pharmacol Ther Toxicol (1984) 22, 626–9.
2. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
3. Moore L, Kramer A, Swites B, Kramer P, Tu J. Effect of cimetidine and antacid on the kinetics of the active diacid of fosinopril in healthy subjects. J Clin Pharmacol (1988) 28, 946.
4. Todd PA, Benfield P. Ramipril. A review of its pharmacological properties and therapeutic efficacy in cardiovascular disorders. Drugs (1990) 39, 110–35.
5. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Accupro (Quinapril hydrochloride). Pfizer Ltd. UK Summary of product characteristics, March 2007.
7. Gopten (Trandolapril). Abbott Laboratories Ltd. UK Summary of product characteristics, May 2007.
ACE inhibitors + Antipsychotics
Marked postural hypotension occurred in a patient given chlorpromazine and captopril. The hypotensive adverse effects of antipsychotics such as the phenothiazines may be additive with the effects of ACE inhibitors.
Clinical evidence, mechanism, importance and management A patient fainted and developed marked postural hypotension (standing blood pressure 66/48 mmHg) when given captopril 6.25 mg twice daily and chlorpromazine 200 mg three times daily. He had previously taken chlorpromazine with nadolol, prazosin and hydrochlorothiazide without any problems, although his blood pressure was poorly controlled on these drugs. Since the patient’s blood pressure was quite elevated when taking chlorpromazine or captopril alone, there appeared to be a synergistic hypotensive effect between the two drugs.1
The manufacturers of several ACE inhibitors warn that ACE inhibitors may enhance the hypotensive effects of certain antipsychotics, and that postural hypotension may occur. Some of these warnings are based, not unreasonably, on the adverse reactions seen with other ACE inhibitors or antihypertensives, but not necessarily on direct observations.2 If postural hypotension occurs warn patients to lay down and elevate their legs if they feel faint or dizzy, and, when recovered, to get up slowly. Dosage adjustments may be necessary to accommodate this interaction.
1. White WB. Hypotension with postural syncope secondary to the combination of chlorpromazine and captopril. Arch Intern Med (1986) 146, 1833–4.
2. Knoll Ltd. Personal communication,1993.
ACE inhibitors + Aprotinin
Aprotinin suppressed the hypotensive action of captopril and enalapril in rats.
Clinical evidence, mechanism, importance and management
A study in spontaneously hypertensive rats found that aprotinin suppressed the hypotensive responses of captopril and enalapril.1 Aprotinin is a proteolytic enzyme inhibitor that has many actions including antagonism of the kallikrein-kinin system, which in turn affects bradykinins and renin. It would therefore be expected to have complex interactions with the ACE inhibitors,2 which also affect these proteins. However, there does not appear to be any evidence to suggest that this theoretical interaction is of clinical relevance.
1. Sharma JN, Amrah SS, Noor AR. Suppression of hypotensive responses of captopril and enalapril by the kallikrein inhibitor aprotinin in spontaneously hypertensive rats. Pharmacology (1995) 50, 363–9.
2. Waxler B, Rabito SF. Aprotinin: a serine protease inhibitor with therapeutic actions: its interaction with ACE inhibitors. Curr Pharm Des (2003) 9, 777–87.
ACE inhibitors + Aspirin
The antihypertensive efficacy of captopril and enalapril may be reduced by high-dose aspirin in about 50% of patients. Low-dose aspirin (less than or equal to 100 mg daily) appears to have little effect. It is unclear whether aspirin attenuates the benefits of ACE inhibitors in heart failure. The likelihood of an interaction may depend on disease state and its severity.
Renal failure has been reported in a patient taking captopril and aspirin.
Clinical evidence
A. Effects on blood pressure
(a) Captopril
Aspirin 600 mg every 6 hours for 5 doses did not significantly alter the blood pressure response to a single 25 to 100-mg dose of captopril in 8 patients with essential hypertension. However, the prostaglandin response to captopril was blocked in 4 of the 8, and in these patients, the blood pressure
response to captopril was blunted.1 In another study, aspirin 75 mg daily did not alter the antihypertensive effects of captopril 25 mg twice daily in 15 patients with hypertension.2
(b) Enalapril
Two groups of 26 patients, one with mild to moderate hypertension taking enalapril 20 mg twice daily and the other with severe primary hypertension taking enalapril 20 mg twice daily (with nifedipine 30 mg and atenolol 50 mg daily), were given test doses of aspirin 100 and 300 mg daily for 5 days. The 100-mg dose of aspirin did not alter the efficacy of the antihypertensive drugs, but the 300-mg dose reduced the antihypertensive efficacy in about half the patients in both groups. In these patients, the antihypertensive effects were diminished by 63% in those with mild to moderate hypertension and by 91% in those with severe hypertension.3 In contrast, another study in 7 patients with hypertension taking enalapril (mean daily dose 12.9 mg) found that aspirin 81 mg or 325 mg daily for 2 weeks did not have any significant effect on blood pressure.4 A further study in 18 patients also found that aspirin 100 mg daily for 2 weeks did not alter the antihypertensive effect of enalapril 20 or 40 mg daily.5
(c) Unspecified ACE inhibitors
In a randomised study, the use of low-dose aspirin 100 mg daily for 3 months did not alter blood pressure control in patients taking calciumchannel blockers or ACE inhibitors, when compared with placebo.6 Similarly, in a re-analysis of data from the Hypertension Optimal Treatment (HOT) study, long-term low-dose aspirin 75 mg daily did not interfere with the blood pressure-lowering effects of the antihypertensive drugs studied, when compared with placebo. Of 18 790 treated hypertensive patients, about 82% received a calcium-channel blocker, usually felodipine alone or in combination, and 41% received an ACE inhibitor, usually in combination with felodipine.7
B. Effects in coronary artery disease and heart failure
Various pharmacological studies have looked at the short-term effects of the combination of ACE inhibitors and aspirin on haemodynamic parameters.
In one study in 40 patients with decompensated heart failure, aspirin 300 mg given on the first day and 100 mg daily thereafter antagonised the short-term haemodynamic effects of captopril 50 mg given every 8 hours for 4 days. The captopril-induced increase in cardiac index and the reduction in peripheral vascular resistance and pulmonary wedge pressure were all abolished.8 In another study, in 15 patients with chronic heart failure receiving treatment with ACE inhibitors (mainly enalapril 10 mg twice daily), aspirin in doses as low as 75 mg impaired vasodilatation induced by arachidonic acid.9 In yet another study, aspirin 325 mg daily worsened pulmonary diffusion capacity and made the ventilatory response to exercise less effective in patients taking enalapril 10 mg twice daily, but did not exert this effect in the absence of ACE inhibitors.10 However, results from studies are inconsistent. In a review,11 five of 7 studies reported aspirin did not alter the haemodynamic effects of ACE inhibitors whereas the remaining two did. In one of these studies showing an adverse interaction between aspirin and enalapril, ticlopidine did not interact with enalapril.12
A number of large clinical studies of ACE inhibitors, mostly post-myocardial infarction, have been re-examined to see if there was a difference in outcome between those receiving aspirin at baseline, and those not. The results are summarised in ‘Table 2.2’, (p.15). However, in addition to the problems of retrospective analysis of non-randomised parameters, the studies vary in the initiation and duration of aspirin and ACE inhibitor treatment and the length of follow-up, the degree of heart failure or ischaemia, the prognosis of the patients, and the final end point (whether compared with placebo or with the benefits of aspirin or ACE inhibitors). The conclusions are therefore conflicting, and, although two meta-analyses of these studies found no interaction, an editorial13 disputes the findings of one of these analyses.14 In addition to these sub-group analyses, there have been a number of retrospective cohort studies. A retrospective study involving 576 patients with heart failure requiring hospitalisation, showed a trend towards an increased incidence of early readmissions (within 30 days after discharge) for heart failure among subjects treated with ACE inhibitors and aspirin, compared with those treated with ACE inhibitors without aspirin (16% versus 10%). In patients without coronary artery disease the increase in readmissions was statistically significant (23% versus 10%).15 However, long-term survival in heart failure was not affected by the use of aspirin with ACE inhibitors. Furthermore, among patients with coronary artery disease there was a trend towards improvement in mortality in patients treated with the combination, compared with ACE inhibitor without aspirin (40% versus 56%).16 Similarly, a lack of adverse interaction was found in a retrospective study involving 14 129 elderly patients who survived a hospitalisation for acute myocardial infarction. However, the added benefit of the combination over patients who received either aspirin or ACE inhibitors alone was not statistically significant.17 Similarly, in another cohort of patients discharged after first hospitalisation for heart failure, there was no increase in mortality rates or readmission rates in those taking aspirin and ACE inhibitors.18 In another retrospective analysis in patients with stable left ventricular systolic dysfunction, no decrease in survival was seen in patients receiving ACE inhibitors, when comparing those also receiving aspirin (mean dose 183 mg daily, 74% 200 mg or less) and those not.19 Conversely, another study found that, compared to patients not taking aspirin, the use of high-dose aspirin (325 mg daily or more) with an ACE inhibitor was associated with a small but statistically significant 3% increase in the risk of death, whereas low-dose aspirin (160 mg daily or less) was not.20
C. Effects on renal function
Acute renal failure developed in a woman taking captopril when she started to take aspirin for arthritis. Renal function improved when both were stopped.21 However, in a re-analysis of data from the Hypertension Optimal Treatment (HOT) study, long-term low-dose aspirin 75 mg daily had no effect on changes in serum creatinine, estimated creatinine clearance or the number of patients developing renal impairment, when compared with placebo. Of 18 790 treated hypertensive patients, 41% received an ACE inhibitor.7
D. Pharmacokinetic studies
A single-dose study in 12 healthy subjects found that the pharmacokinetics of benazepril 20 mg and aspirin 325 mg were not affected by concurrent use.22
Mechanism
Some, but not all the evidence suggests that prostaglandins may be involved in the hypotensive action of ACE inhibitors, and that aspirin, by inhibiting prostaglandin synthesis, may partially antagonise the effect of ACE inhibitors on blood pressure. This effect appears to depend on the dose of aspirin and may also be dependent on sodium status and plasma renin, and therefore it does not occur in all patients.
The beneficial effects of ACE inhibitors in heart failure and ischaemic heart disease are thought to be due, in part, to the inhibition of the breakdown of kinins, which are important regulators of prostaglandin and nitric oxide synthesis. Such inhibition promotes vasodilatation and afterload reduction.
Aspirin may block these beneficial effects by inhibiting cyclo-oxygenase (COX) and thus prostaglandin synthesis, causing vasoconstriction, decreased cardiac output and worsening heart failure.11,23
Importance and management
Low-dose aspirin (less than or equal to 100 mg daily) does not alter the antihypertensive efficacy of captopril and enalapril. No special precautions would therefore seem to be required with ACE inhibitors and these low doses of aspirin. A high dose of aspirin (2.4 g daily) has been reported to interact in 50% of patients in a single study. Aspirin 300 mg daily has been reported to interact in about 50% of patients in another study, whereas 325 mg daily did not interact in further study. Thus, at present, it appears that if an ACE inhibitor is used with aspirin in doses higher than 300 mg daily, blood pressure should be monitored more closely, and the ACE inhibitor dosage raised if necessary. Intermittent use of aspirin should be considered as a possible cause of erratic control of blood pressure in patients on ACE inhibitors.
Both ACE inhibitors and aspirin are often taken by patients with coronary artery disease, and ACE inhibitors are used in chronic heart failure, which is often associated with coronary heart disease. The
information about a possible interaction between ACE inhibitors and aspirin in heart failure is conflicting. This may be due to much of the clinical data being obtained from retrospective non-randomised analyses.23 It may also be a factor of different disease states. For example, an interaction may be less likely to be experienced in patients with heart failure of ischaemic aetiology than those with non-ischaemic causes, because of the added benefits of aspirin in ischaemic heart disease.24 The available data, and its implications, have been extensively reviewed and commented on.11,13,23-32 Some commentators have advised that, if possible, aspirin should be avoided in patients requiring long-term treatment for heart failure, particularly if heart failure is severe.13,27 Others suggest avoiding aspirin in heart failure unless there are clear indications, such as atherosclerosis.
11,23,28,29 The use of lower doses of aspirin (80 to 100 mg daily rather than greater than or equal to 325 mg daily) in those with heart failure taking ACE inhibitors has also been suggested.24,25,28 US guidelines from 2005 on chronic heart failure33 state that, “Many physicians believe the data justify prescribing aspirin and ACE inhibitors together when there is an indication for use of aspirin,” while recognising that not all physicians agree. The guidelines say that further study is needed. European guidelines state that there is little evidence to support using ACE inhibitors and aspirin together in heart failure. The guidelines say aspirin can be used as prophylaxis after prior myocardial infarction, but that it should be avoided in patients with recurrent hospitalisation for worsening heart failure.34 NICE guidelines in the UK make no comment about the combination of ACE inhibitors and aspirin. They say that all patients with heart failure due to left ventricular systolic dysfunction should be considered for treatment with an ACE inhibitor, and that aspirin (75 to 150 mg once daily) should be prescribed for patients with the combination of heart failure and atherosclerotic arterial disease (including coronary heart disease).35 Data from ongoing randomised studies may provide further insight. Until these are available, combined low-dose aspirin and ACE inhibitors may continue to be used where there is a clear indication for both.
An increased risk of deterioration in renal function or acute renal failure appears to occur rarely with the combination of aspirin and ACE inhibitors. The routine monitoring of renal function, which is advised with ACE inhibitors, should be sufficient to detect any interaction.
1. Moore TJ, Crantz FR, Hollenberg NK, Koletsky RJ, Leboff MS, Swartz SL, Levine L, Podolsky S, Dluhy RG, Williams GH. Contribution of prostaglandins to the antihypertensive action of captopril in essential hypertension. Hypertension (1981) 3, 168–73.
2. Smith SR, Coffman TM, Svetkey LP. Effect of low-dose aspirin on thromboxane production and the antihypertensive effect of captopril. J Am Soc Nephrol (1993) 4, 1133–9.
3. Guazzi MD, Campodonico J, Celeste F, Guazzi M, Santambrogio G, Rossi M, Trabattoni D, Alimento M. Antihypertensive efficacy of angiotensin converting enzyme inhibition and aspirin counteraction. Clin Pharmacol Ther (1998) 63, 79–86.
4. Nawarskas JJ, Townsend RR, Cirigliano MD, Spinler SA. Effect of aspirin on blood pressure in hypertensive patients taking enalapril or losartan. Am J Hypertens (1999) 12, 784–9.
5. Polónia J, Boaventura I, Gama G, Camões I, Bernardo F, Andrade P, Nunes JP, Brandão F, Cerqueira-Gomes M. Influence of non-steroidal anti-inflammatory drugs on renal function and 24 h ambulatory blood pressure-reducing effects of enalapril and nifedipine gastrointestinal therapeutic system in hypertensive patients. J Hypertens (1995) 13, 925–31
6. Avanzini F, Palumbo G, Alli C, Roncaglioni MC, Ronchi E, Cristofari M, Capra A, Rossi S, Nosotti L, Costantini C, Pietrofeso R. Collaborative Group of the Primary Prevention Project (PPP)–Hypertension study. Effects of low-dose aspirin on clinic and ambulatory blood pressure in treated hypertensive patients. Am J Hypertens (2000) 13, 611–16.
7. Zanchetti A, Hansson L, Leonetti G, Rahn K-H, Ruilope L, Warnold I, Wedel H. Low-dose aspirin does not interfere with the blood pressure-lowering effects of antihypertensive therapy. J Hypertens (2002) 20, 1015–22.
8. Viecili PR, Pamplona D, Park M, Silva SR, Ramires JAF, da Luz PL. Antagonism of the acute hemodynamic effects of captopril in decompensated congestive heart failure by aspirin administration. Braz J Med Biol Res (2003) 36, 771–80.
9. Davie AP, Love MP, McMurray JJV. Even low-dose aspirin inhibits arachidonic acid-induced vasodilation in heart failure. Clin Pharmacol Ther (2000) 67, 530–7.
10. Guazzi M, Pontone G, Agostoni P. Aspirin worsens exercise performance and pulmonary gas exchange in patients with heart failure who are taking angiotensin-converting enzyme inhibitors. Am Heart J (1999) 138, 254–60.
11. Mahé I, Meune C, Diemer M, Caulin C, Bergmann J-F. Interaction between aspirin and ACE inhibitors in patients with heart failure. Drug Safety (2001) 24, 167–82.
12. Spaulding C, Charbonnier B, Cohen-Solal A, Juillière Y, Kromer EP, Benhamda K, Cador R, Weber S. Acute hemodynamic interaction of aspirin and ticlopidine with enalapril. Results of a double-blind, randomized comparative trial. Circulation (1998) 98, 757–65.
13. Hall D. The aspirin—angiotensin-converting enzyme inhibitor tradeoff: to halve and halve not. J Am Coll Cardiol (2000) 35, 1808–12.
14. Latini R, Tognoni G, Maggioni AP, Baigent C, Braunwald E, Chen Z-M, Collins R, Flather M, Franzosi MG, Kjekshus J, Køber L, Liu L-S, Peto R, Pfeffer M, Pizzetti F, Santoro E, Sleight P, Swedberg K, Tavazzi L, Wang W, Yusuf S, on behalf of the Angiotensin-converting Enzyme Inhibitor Myocardial Infarction Collaborative Group. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for acute myocardial infarction are similar in the presence and absence of aspirin. Systematic overview of individual data from 96,712 randomized patients. J Am Coll Cardiol (2000) 35 1801–7.
15. Harjai KJ, Nunez E, Turgut T, Newman J. Effect of combined aspirin and angiotensin-converting
enzyme inhibitor therapy versus angiotensin-converting enzyme inhibitor therapy alone on readmission rates in heart failure. Am J Cardiol (2001) 87, 483–7.
16. Harjai KJ, Solis S, Prasad A, Loupe J. Use of aspirin in conjunction with angiotensin-converting
enzyme inhibitors does not worsen long-term survival in heart failure. Int J Cardiol (2003) 88, 207–14.
17. Krumholz HM, Chen Y-T, Wang Y, Radford MJ. Aspirin and angiotensin-converting enzyme inhibitors among elderly survivors of hospitalization for an acute myocardial infarction.
Arch Intern Med (2001) 161, 538–44.
18. McAlister FA, Ghali WA, Gong Y, Fang J, Armstrong PW, Tu JV. Aspirin use and outcomes in a community-based cohort of 7352 patients discharged after first hospitalization for heart failure. Circulation (2006) 113, 2572–8.
19. Aumégeat V, Lamblin N, de Groote P, Mc Fadden EP, Millaire A, Bauters C, Lablanche JM. Aspirin does not adversely affect survival in patients with stable congestive heart failure treated with angiotensin-converting enzyme inhibitors. Chest (2003) 124, 1250–8.
20. Guazzi M, Brambilla R, Rèina G, Tumminello G, Guazzi MD. Aspirin-angiotensin-converting enzyme inhibitor coadministration and mortality in patients with heart failure: a dose-related adverse effect of aspirin. Arch Intern Med (2003) 163, 1574–9.
21. Seelig CB, Maloley PA, Campbell JR. Nephrotoxicity associated with concomitant ACE inhibitor
and NSAID therapy. South Med J (1990) 83, 1144–8.
22. Sioufi A, Pommier F, Gauducheau N, Godbillon J, Choi L, John V. The absence of a pharmacokinetic
interaction between aspirin and the angiotensin-converting enzyme inhibitor
benazepril in healthy volunteers. Biopharm Drug Dispos (1994) 15, 451–61.
23. Massie BM, Teerlink JR. Interaction between aspirin and angiotensin-converting enzyme inhibitors:
real or imagined. Am J Med (2000) 109, 431–3.
24. Nawarskas JJ, Spinler SA. Update on the interaction between aspirin and angiotensin-converting
enzyme inhibitors. Pharmacotherapy (2000) 20, 698–710.
25. Stys T, Lawson WE, Smaldone GC, Stys A. Does aspirin attenuate the beneficial effects of angiotensin-converting enzyme inhibition in heart failure? Arch Intern Med (2000) 160, 1409–13.
26. Barbash IM, Goldbourt U, Gottlieb S, Behar S, Leor J. Possible interaction between aspirin and ACE inhibitors: update on unresolved controversy. Congest Heart Fail (2000) 6, 313–18.
27. Cleland JGF, John J, Houghton T. Does aspirin attenuate the effect of angiotensin-converting enzyme inhibitors in hypertension or heart failure? Curr Opin Nephrol Hypertens (2001) 10, 625–31.
28. Peterson JG, Lauer MS. Using aspirin and ACE inhibitors in combination: why the hullabaloo? Cleve Clin J Med (2001) 68, 569–74.
29. Olson KL. Combined aspirin/ACE inhibitor treatment for CHF. Ann Pharmacother (2001) 35, 1653–8.
30. Park MH. Should aspirin be used with angiotensin-converting enzyme inhibitors in patients with chronic heart failure? Congest Heart Fail (2003) 9, 206–11.
31. Konstam MA. Aspirin and heart failure: square evidence meets a round patient. Congest Heart Fail (2003) 9, 203–5.
32. Brunner-La Rocca HP. Interaction of angiotensin-converting enzyme inhibition and aspirin in congestive heart failure: long controversy finally resolved? Chest (2003) 124, 1192–4.
33. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation (2005) 112, e154–235. Available at: http://circ.ahajournals.org/cgi/content/full/ 112/12/e154 (accessed 14/08/07).
34. Swedberg K, Cleland J, Dargie H, Drexler H, Follath F, Komajda M, Tavazzi L, Smiseth OA, Gavazzi A, Haverich A, Hoes A, Jaarsma T, Korewicki J, Lévy S, Linde C, Lopez-Sendon JL, Nieminen MS, Piérard L, Remme WJ; The Task Force for the Diagnosis and Treatment of CHF of the European Society of Cardiology. Guidelines for the diagnosis and treatment of chronic heart failure: full text (update 2005): The Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology. Eur Heart J (2005) 26, 1115–40. Available at: http://www.escardio.org/NR/rdonlyres/8A2848B4-5DEB-41B9-9A0A-5B5A90494B64/0/ CHFFullTextehi205FVFW170505.pdf (accessed 14/08/07).
35. National Institute for Clinical Excellence. Chronic heart failure: management of chronic heart failure in adults in primary and secondary care (issued July 2003). Available at http://www.nice.org.uk/pdf/CG5NICEguideline.pdf (accessed 14/08/07).


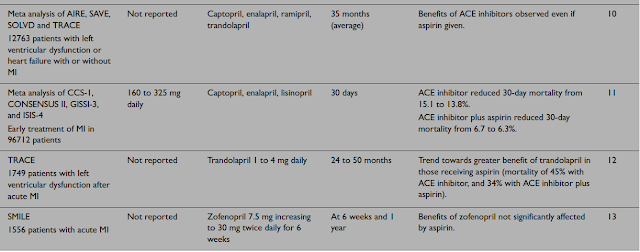

Clinical evidence
(a) Anaemia
Nine out of 11 kidney transplant patients taking ACE inhibitors (enalapril or captopril) had a fall in their haematocrit from 34% to 27%, and a fall in their haemoglobin from 11.6 g/dL to 9.5 g/dL when ciclosporin was replaced by azathioprine. Two patients were switched back to ciclosporin, and had a prompt rise in their haematocrit. Another 10 patients taking both drugs similarly developed a degree of anaemia, when compared with 10 others not taking an ACE inhibitor (haematocrit of 33% compared with 41%, and a haemoglobin of 11.5 g/dL compared with 13.9 g/dL).1 A later study by the same group of workers (again in patients taking enalapril or captopril) confirmed these findings: however, no pharmacokinetic interaction was found between enalapril and azathioprine.2
(b) Leucopenia
A patient whose white cell count fell sharply when taking both captopril 50 mg daily and azathioprine 150 mg daily, did not develop leucopenia when each drug was given separately.3 Another patient who was given captopril (increased to 475 mg daily [sic] then reduced to 100 mg daily) immediately after discontinuing azathioprine, developed leucopenia. She was later successfully treated with captopril 4 to 6 mg daily [sic].4 Other patients have similarly shown leucopenia when given both drugs;5,6 in one case this did not recur when the patient was rechallenged with captopril
alone (at a lower dose).6
Mechanism
The anaemia appears to be due to suppression of erythropoietin by the ACE inhibitors, and azathioprine may cause patients to be more susceptible to this effect.2 The cause of the leucopenia is unknown. It may just be due to the additive effects of both drugs.
Importance and management
Anaemia caused by captopril and enalapril has been seen in kidney transplant patients and in dialysis patients (see ‘ACE inhibitors and Angiotensin II receptor antagonists + Epoetin’, p.25). The evidence that this effect can be potentiated by azathioprine is limited, but it would be prudent to monitor well if these drugs are used together.
The evidence that the concurrent use of ACE inhibitors and azathioprine increases the risk of leucopenia is also limited. However, the UK manufacturer of captopril recommends that captopril should be used with extreme caution in patients receiving immunosuppressants, especially if there is renal impairment. They advise that in such patients differential white blood cell counts should be performed before starting captopril, then every 2 weeks in the first 3 months of treatment, and periodically thereafter.
7 The UK manufacturers of a number of other ACE inhibitors also state in their prescribing information that the use of ACE inhibitors with cytostatic or immunosuppressive drugs may lead to an increased risk of leucopenia.
For other potential interactions with ACE inhibitors that might lead to an increased risk of leucopenia, see also ‘ACE inhibitors + Allopurinol’, p.13, and ‘ACE inhibitors + Procainamide’, p.33.
1. Gossmann J, Kachel H-G, Schoeppe W, Scheuermann E-H. Anemia in renal transplant recipients caused by concomitant therapy with azathioprine and angiotensin-converting enzyme inhibitors.
Transplantation (1993) 56, 585–9.
2. Gossmann J, Thürmann P, Bachmann T, Weller S, Kachel H-G, Schoeppe W, Scheuermann E-H. Mechanism of angiotensin converting enzyme inhibitor-related anemia in renal transplant recipients. Kidney Int (1996) 50, 973–8.
3. Kirchertz EJ, Gröne HJ, Rieger J, Hölscher M, Scheler F. Successful low dose captopril rechallenge following drug-induced leucopenia. Lancet (1981) i, 1363.
4. Case DB, Whitman HH, Laragh JH, Spiera H. Successful low dose captopril rechallenge following drug-induced leucopenia. Lancet (1981) i, 1362–3.
5. Elijovisch F, Krakoff LR. Captopril associated granulocytopenia in hypertension after renal transplantation. Lancet (1980), i, 927–8.
Clinical evidence
(a) Atenolol
In a double-blind, crossover study in hypertensive subjects, the combination of atenolol 50 mg once daily and enalapril 20 mg once daily increased the hypotensive effect of either drug alone, but the effect was 30 to 50% less than additive.1
(b) Bisoprolol
In a single-dose, placebo-controlled, crossover study in 16 healthy men, bisoprolol 5 mg given with imidapril 10 mg did not significantly influence the pharmacokinetics of its active metabolite imidaprilat, and the pharmacodynamic effects, including blood pressure and heart rate reductions, were mainly additive.2
(c) Propranolol
Propranolol 80 mg three times daily did not affect the pharmacokinetics of a single 20-mg dose of quinapril in 10 healthy subjects.3 The pharmacokinetics of ramipril 5 mg daily were unaffected by propranolol 40 mg twice daily.4 Similarly, the manufacturer of fosinopril reports that the bioavailability of fosinoprilat, its active metabolite, was not altered by propranolol.
5,6 Another study found no significant pharmacokinetic interaction between cilazapril 2.5 mg daily and propranolol 120 mg daily in healthy subjects, but the reductions in blood pressure were about doubled and long-lasting in healthy subjects and in patients with hypertension.7,8
Mechanism, importance and management
Both ACE inhibitors and beta blockers lower blood pressure by different mechanisms, and therefore the enhanced blood pressure-lowering effects of the combination would be expected. No pharmacokinetic interactions have been demonstrated. The combination of an ACE inhibitor and a beta blocker is clinically useful in a number of cardiovascular disorders.
1. Wing LMH, Chalmers JP, West MJ, Russell AE, Morris MJ, Cain MD, Bune AJC, Southgate DO. Enalapril and atenolol in essential hypertension: attenuation of hypotensive effects in combination. Clin Exp Hypertens (1988) 10, 119–33.
2. Breithaupt-Grögler K, Ungethüm W, Meurer-Witt B, Belz GG. Pharmacokinetic and dynamic interactions of the angiotensin-converting enzyme inhibitor imidapril with hydrochlorothiazide, bisoprolol and nilvadipine. Eur J Clin Pharmacol (2001) 57, 275–84.
3. Horvath AM, Pilon D, Caillé G, Colburn WA, Ferry JJ, Frank GJ, Lacasse Y, Olson SC. Multiple-dose propranolol administration does not influence the single dose pharmacokinetics of quinapril and its active metabolite (quinaprilat). Biopharm Drug Dispos (1990) 11, 191–6.
4. van Griensven JMT, Seibert-Grafe M, Schoemaker HC, Frölich M, Cohen AF. The pharmacokinetic and pharmacodynamic interactions of ramipril with propranolol. Eur J Clin Pharmacol (1993) 45, 255–60.
5. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
7. Belz GG, Essig J, Kleinbloesem CH, Hoogkamer JFW, Wiegand UW, Wellstein A. Interactions between cilazapril and propranolol in man; plasma drug concentrations, hormone and enzyme responses, haemodynamics, agonist dose-effect curves and baroreceptor reflex. Br J Clin Pharmacol (1988) 26, 547–56.
8. Belz GG, Essig J, Erb K, Breithaupt K, Hoogkamer JFW, Kneer J, Kleinbloesem CH. Pharmacokinetic and pharmacodynamic interactions between the ACE inhibitor cilazapril and β-adrenoceptor antagonist propranolol in healthy subjects and in hypertensive patients. Br J Clin Pharmacol (1989) 27, 317S–322S.
(a) Amlodipine
A study in 12 healthy subjects indicated that there was no pharmacokinetic interaction between single doses of amlodipine 5 mg and benazepril 10 mg.1
(b) Felodipine
No pharmacokinetic interaction occurred between single doses of felodipine 10 mg and ramipril 5 mg in healthy subjects. The blood pressure-lowering effect of the combination was greater, and ramipril attenuated the reflex tachycardia caused by felodipine.2
(c) Manidipine
In a single-dose crossover study in 18 healthy subjects, the concurrent use of manidipine 10 mg and delapril 30 mg did not significantly alter the pharmacokinetics of either drug or their main metabolites.3
(d) Nicardipine
In a study in 12 patients with hypertension taking enalapril 20 mg daily, the addition of nicardipine 30 mg three times daily for 2 weeks did not alter the pharmacokinetics of enalapril.4 The manufacturer of spirapril briefly noted in a review that the concurrent use of spirapril and nicardipine
increased spirapril plasma levels by about 25% and those of its active metabolite, spiraprilat, by about 45%. The bioavailability of nicardipine was reduced by 30%. It was assumed that the interaction took
place at the absorption site. However, the changes were not considered clinically relevant.5
(e) Nifedipine
No evidence of either a pharmacokinetic or adverse pharmacodynamic interaction was seen in 12 healthy subjects given single doses of nifedipine retard 20 mg and lisinopril 20 mg; the effects on blood pressure were additive. 6 Similarly, there was no pharmacokinetic interaction between single doses of slow-release nifedipine 20 mg and benazepril 10 mg in healthy subjects; the effects on blood pressure were additive and the tachycardic effect of nifedipine was attenuated by benazepril.7 The manufacturer of fosinopril notes that the bioavailability of fosinoprilat, the active metabolite, was not altered by nifedipine.8,9 Similarly, the manufacturer of moexipril notes that no clinically important pharmacokinetic interaction occurred with nifedipine in healthy subjects.10
(f) Nilvadipine
In a single-dose, placebo-controlled, crossover study in 16 healthy subjects, no pharmacokinetic interaction occurred between nilvadipine 8 mg and imidapril 10 mg, and the pharmacodynamic effects, including the reduction in blood pressure and the decrease in total peripheral resistance,
were mostly additive.11
No pharmacokinetic interactions are expected. Enhanced blood pressurelowering effects occur, as would be expected.
No important pharmacokinetic interactions have been demonstrated. The combination of an ACE inhibitor and a dihydropyridine calcium-channel blocker is clinically useful in the treatment of hypertension. A number of products combining an ACE inhibitor with a calcium-channel blocker are available. It is generally advised that these combination products are only used in patients who have already been stabilised on the individual components in the same proportions.
1. Sun JX, Cipriano A, Chan K, John VA. Pharmacokinetic interaction study between benazepril and amlodipine in healthy subjects. Eur J Clin Pharmacol (1994) 47, 285–9.
2. Bainbridge AD, MacFadyen RJ, Lees KR, Reid JL. A study of the acute pharmacodynamic interaction of ramipril and felodipine in normotensive subjects. Br J Clin Pharmacol (1991) 31, 148–53.
3. Stockis A, Gengler C, Goethals F, Jeanbaptiste B, Lens S, Poli G, Acerbi D. Single oral dose pharmacokinetic interaction study of manidipine and delapril in healthy volunteers. Arzneimittelforschung (2003) 53, 627–34.
4. Donnelly R, Elliott HL, Reid JL. Nicardipine combined with enalapril in patients with essential hypertension. Br J Clin Pharmacol (1986) 22, 283S–287S.
5. Grass P, Gerbeau C, Kutz K. Spirapril: pharmacokinetic properties and drug interactions. Blood Pressure (1994) 3 (Suppl 2), 7–13.
6. Lees KR, Reid JL. Lisinopril and nifedipine: no acute interaction in normotensives. Br J Clin Pharmacol (1988) 25, 307–13.
Clinical evidence, mechanism, importance and management
A 53-year-old woman who had been taking an unnamed ACE inhibitor for several years, complained of cough each time she applied Axsain, a cream containing capsaicin 0.075%, to her lower extremities. Whether this reaction would have occurred without the ACE inhibitor was not determined,1 but cough is a recognised adverse effect of ACE inhibitors and pre-treatment with an ACE inhibitor has been shown to enhance the cough caused by inhaled capsaicin.1 This potential interaction is probably of little general clinical importance.
1. Hakas JF. Topical capsaicin induces cough in patient receiving ACE inhibitor. Ann Allergy (1990) 65, 322.
(a) Albumin
A woman taking enalapril 10 mg in the morning, underwent surgery for groin lymph node resection under spinal and general anaesthesia. When she was given a rapid infusion of 500 mL of the albumin solution, stable plasma protein solution (SPPS, Commonwealth Serum Laboratories, Melbourne, Australia), her pulse rose to 90 to 100 bpm and systolic blood pressure fell from 100 to 60 mmHg and a red flush was noted on all exposed skin. The blood pressure was controlled at 90 to 95 mmHg with metaraminol 4.5 mg, given over 10 minutes. When the SPPS was finished, the blood pressure and pulse rate spontaneously restabilised.1 SPPS is a 5% plasma protein solution prepared by the cold ethanol fractionation process and pasteurisation from human plasma (volunteer donors). It contains sodium octanoate as a stabiliser.1 Two very similar cases have been recorded in patients taking enalapril when given SPPS.2,3 The manufacturer of SPPS notes that captopril has also been involved in this hypotensive interaction A 20-month-old infant taking captopril was haemodynamically stable for 35 minutes after induction of anaesthesia while awaiting a donor kidney, but then developed hypotension after a bolus dose of 20 mL of albumin 4% (Albumex) was given. This was reversed with dopamine infusion.5
(b) Gelatin-based colloids
A report describes 3 cases of severe hypotension in patients taking ACE inhibitors (lisinopril, enalapril) while undergoing joint replacement surgery, and after they had been given a gelatin-based plasma expander (Gelofusin), which contains 4% succinylated gelatin in saline. The hypotension was resistant to ephedrine and methoxamine, and responded to adrenaline or dobutamine, which was required for 24 hours and 3 days in two cases. Anaphylactoid reactions were excluded as a cause of the hypotension.6 In another similar case, a patient taking cilazapril developed hypotension refractory to sodium chloride 0.9% after induction of anaesthesia, and this worsened when a gelatin-type colloid (Gelafundina) was given.7
Importance and management
The interaction with SPPS would appear to be established and of clinical importance, and would apply to all ACE inhibitors. The author of one report suggested that if rapid expansion of intravascular volume is needed in patients taking ACE inhibitors, an artificial colloid might be a safer choice than SPPS.1 The manufacturer of SPPS also recommended using an alternative plasma volume expander, including other albumin solutions.4 It should be noted that following these reports SPPS was withdrawn from the Australasian market.9 However, note that a case has also occurred with albumin 4%, and cases have also been attributed to synthetic colloid solutions containing gelatin. It may be that this is just an unpredictable effect of colloids in patients taking ACE inhibitors. See also ‘Anaesthetics, general + Antihypertensives’, p.94, for discussion of the marked hypotension sometimes seen during induction of anaesthesia in patients taking ACE inhibitors.
1. McKenzie AJ. Possible interaction between SPPS and enalapril. Anaesth Intensive Care (1990) 18, 124–6.
2. Young K. Enalapril and SPPS. Anaesth Intensive Care (1990) 18, 583.
3. Young K. Hypotension from the interaction of ACE inhibitors with stable plasma protein solution. Anaesthesia (1993) 48, 356.
4. Schiff P. SPPS, hypotension and ACE inhibitors. Med J Aust (1992) 156, 363.
5. Fong SY, Hansen TG. Perioperative hypotension following plasma volume expansion with albumin in an angiotensin-converting enzyme inhibited infant. Br J Anaesth (2000) 84, 537–8.
6. Powell CG, Unsworth DJ, McVey FK. Severe hypotension associated with angiotensin-converting enzyme inhibition in anaesthesia. Anaesth Intensive Care (1998) 26, 107–9.
7. Barber L, Barrio J, de Rojas MD, Ibañez F, Añó C, Alepuz R, Montero R. Hipotensión refractaria y sostenida durante una anestesia general asociada al tratamiento crónico con inhibidores de la enzima conversiva de la angiotensina. Rev Esp Anestesiol Reanim. (2001) 48, 34–7.
8. Bönner G, Preis S, Schunk U, Toussaint C, Kaufmann W. Hemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans. J Cardiovasc Pharmacol (1990) 15 (Suppl 6), S46–S56.
9. McKenzie AJ. ACE inhibitors, colloid infusions and anaesthesia. Anaesth Intensive Care (1998) 26, 330.



J Clin Pharmacol (2003) 43, 443–69.



(d) Genetic factors in drug metabolism
An increased understanding of genetics has shown that some of the cytochrome P450 isoenzymes are subject to ‘genetic polymorphism’, which simply means that some of the population have a variant of the isoenzyme with different (usually poor) activity. The best known example is CYP2D6, for which a small proportion of the population have the variant with low activity and are described as being poor or slow metabolisers (about 5 to 10% in white Caucasians, 0 to 2% in Asians and black people).
Which group any particular individual falls into is genetically determined.
The majority who possess the isoenzyme are called ‘fast or extensive metabolisers’. It is possible to find out which group any particular individual falls into by looking at the way a single dose of a test or ‘probe’ drug is metabolised. This varying ability to metabolise certain drugs may explain why some patients develop toxicity when given an interacting drug while others remain symptom free. CYP2D6, CYP2C9 and CYP2C19 also show polymorphism, whereas CYP3A4 does not, although there is still some broad variation in the population without there being distinct groups. The effects of CYP2C19 polymorphism are discussed in more detail in ‘Gastrointestinal drugs’, (p.960). At present, genotyping of cytochrome P450 isoenzymes is primarily a research tool and is not used clinically. In the future, it may become standard clinical practice and may be used to individualise drug therapy.1
(e) Cytochrome P450 isoenzymes and predicting drug interactions
It is interesting to know which particular isoenzyme is responsible for the metabolism of drugs because by doing in vitro tests with human liver enzymes it is often possible to explain why and how some drugs interact. For example, ciclosporin is metabolised by CYP3A4, and we know that rifampicin (rifampin) is a potent inducer of this isoenzyme, whereas ketoconazole inhibits its activity, so that it comes as no surprise that rifampicin reduces the effects of ciclosporin and ketoconazole increases it. What is very much more important than retrospectively finding out why two drugs interact, is the knowledge such in vitro tests can provide about forecasting which other drugs may possibly also interact. This may reduce the numbers of expensive clinical studies in subjects and patients and avoids waiting until significant drug interactions are observed in clinical use. A lot of effort is being put into this area of drug development.2-6 However, at present such prediction is, like weather forecasting, still a somewhat hit-and-miss business because we do not know all of the factors that may modify or interfere with metabolism. It is far too simplistic to think that we have all the answers just because we know which liver isoenzymes are concerned with the metabolism of a particular drug, but it is a very good start.‘Table 1.2’, (p.4), ‘Table 1.3’, (p.6), ‘Table 1.4’, (p.6) are lists of drugs that are inhibitors, inducers, or substrates of the clinically important cytochrome P450 isoenzymes, and each drug has a cross reference to a monograph describing a drug interaction thought to occur via that mechanism.
If a new drug is shown to be an inducer, or an inhibitor, and/or a substrate of a given isoenzyme, these tables could be used to predict likely drug interactions. However, what may happen in vitro may not necessarily work in clinical practice because all of the many variables which can come into play are not known (such as how much of the enzyme is available, the concentration of the drug at the site of metabolism, and the affinity of the drug for the enzyme). Remember too that some drugs can be metabolised by more than one cytochrome P450 isoenzyme (meaning that this other isoenzyme may be able to ‘pick up’ more metabolism to compensate for the inhibited pathway); some drugs (and their metabolites) can both induce a particular isoenzyme and be metabolised by it; and some drugs (or their metabolites) can inhibit a particular isoenzyme but not be metabolised by it. With so many factors possibly impinging on the outcome of giving two or more drugs together, it is very easy to lose sight of one of the factors (or not even know about it) so that the sum of 2 plus 2 may not turn out to be the 4 that you have predicted.
For example, ritonavir and other protease inhibitors are well known potent inhibitors of CYP3A4, and in clinical use increase the levels of many drugs that are substrates of this isoenzyme. Methadone is a substrate of CYP3A4, and some in vitro data show that ritonavir (predictably) increased methadone levels. However, unexpectedly, in clinical use the protease inhibitors seem to decrease methadone levels, by a yet unknown mechanism (see, ‘Opioids; Methadone + Protease inhibitors’, p.182).
Another factor complicating the understanding of metabolic drug interactions is the finding that there is a large overlap between the inhibitors/inducers and substrates of P-glycoprotein (a ‘drug transporter protein’, (p.8)) and those of CYP3A4. Therefore, both mechanisms may be involved in many of the drug interactions previously thought to be due to effects on CYP3A4.
1. Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA (2001) 286, 2270–79.
2. Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a PhRMA perspective. J Clin Pharmacol (2003) 43, 443–69.
3. Bachmann KA, Ghosh R. The use of in vitro methods to predict in vivo pharmacokinetics and drug interactions. Curr Drug Metab (2001) 2, 299–314.
4. Yao C, Levy RH. Inhibition-based metabolic drug–drug interactions: predictions from in vitro data. J Pharm Sci (2002) 91, 1923–35.
5. Worboys PD, Carlile DJ. Implications and consequences of enzyme induction on preclinical and clinical drug development. Xenobiotica (2001) 31, 539–56.
6. Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Drug metabolism and drug interactions: application and clinical value of in vitro models. Curr Drug Metab (2003) 4, 423–59.
1.4. Drug excretion interactions
With the exception of the inhalation anaesthetics, most drugs are excreted either in the bile or in the urine. Blood entering the kidneys along the renal arteries is, first of all, delivered to the glomeruli of the tubules where molecules small enough to pass through the pores of the glomerular membrane (e.g. water, salts, some drugs) are filtered through into the lumen of the tubules.
Larger molecules, such as plasma proteins, and blood cells are retained within the blood. The blood flow then passes to the remaining parts of the kidney tubules where active energy-using transport systems are able to remove drugs and their metabolites from the blood and secrete them into the tubular filtrate. The renal tubular cells additionally possess active and passive transport systems for the reabsorption of drugs. Interference by drugs with renal tubular fluid pH, with active transport systems and with blood flow to the kidney can alter the excretion of other drugs.

(a) Changes in urinary pH
As with drug absorption in the gut, passive reabsorption of drugs depends upon the extent to which the drug exists in the non-ionised lipid-soluble form, which in its turn depends on its pKa and the pH of the urine. Only the non-ionised form is lipid-soluble and able to diffuse back through the lipid membranes of the tubule cells. Thus at high pH values (alkaline), weakly acid drugs (pKa 3 to 7.5) largely exist as ionised lipid-insoluble molecules, which are unable to diffuse into the tubule cells and will there fore remain in the urine and be removed from the body. The converse will be true for weak bases with pKa values of 7.5 to 10.5. Thus pH changes that increase the amount in the ionised form (alkaline urine for acidic drugs, acid urine for basic drugs) increase the loss of the drug, whereas moving the pH in the opposite direction will increase their retention. ‘Figure 1.4’, (p.7) illustrates the situation with a weakly acidic drug. The clinical significance of this interaction mechanism is small, because although a very large number of drugs are either weak acids or bases, almost all are largely metabolised by the liver to inactive compounds and few are excreted in the urine unchanged. In practice therefore only a handful of drugs seem to be affected by changes in urinary pH (possible exceptions include changes in the excretion of ‘quinidine’, (p.277) or ‘analgesic-dose aspirin’, (p.135), due to alterations in urinary pH caused by antacids, and the increase in the clearance of ‘methotrexate’, (p.654), with urinary alkalinisers). In cases of overdose, deliberate manipulation of urinary pH has been used to increase the removal of drugs such as methotrexate and salicylates.

(b) Changes in active renal tubular excretion
Drugs that use the same active transport systems in the renal tubules can compete with one another for excretion. For example, probenecid reduces the excretion of penicillin and other drugs. With the increasing understanding of drug transporter proteins in the kidneys, it is now known that probenecid inhibits the renal secretion of many other anionic drugs by organic anion transporters (OATs).1 Probenecid possibly also inhibits some of the ABC transporters in the kidneys. The ABC transporter, P-glycoprotein, is also present in the kidneys, and drugs that alter this may alter renal drug elimination. See, ‘Drug transporter proteins’, (p.8), for further discussion. Some examples of drugs that possibly interact by alterations in renal transport are given in ‘Table 1.5’, (see above).
(c) Changes in renal blood flow
The flow of blood through the kidney is partially controlled by the production of renal vasodilatory prostaglandins. If the synthesis of these prostaglandins is inhibited the renal excretion of some drugs may be reduced. An interaction where this is the suggested mechanism is the rise in serum lithium seen with some NSAIDs, see ‘Lithium + NSAIDs’, p.1125.
(d) Biliary excretion and the entero-hepatic shunt
(i) Enterohepatic recirculation
A number of drugs are excreted in the bile, either unchanged or conjugated (e.g. as the glucuronide) to make them more water soluble. Some of the conjugates are metabolised to the parent compound by the gut flora and are then reabsorbed. This recycling process prolongs the stay of the drug within the body, but if the gut flora are diminished by the presence of an antibacterial, the drug is not recycled and is lost more quickly. This may possibly explain the rare failure of the oral contraceptives that can be brought about by the concurrent use of penicillins or tetracyclines, but see
Mechanism in ‘Hormonal contraceptives + Antibacterials; Penicillins’, p.981. Antimicrobial-induced reductions in gut bacteria may reduce the activation of ‘sulfasalazine’, (p.973).
(ii) Drug transporters
Increasing research shows that numerous drug transporter proteins (both from the ABC family and SLC family, see ‘Drug transporter proteins’, (see below)) are involved in the hepatic extraction and secretion of drugs into the bile.2 The relevance of many of these to drug interactions is still unclear, but the bile salt export pump (ABCB11) is known to be inhibited by a variety of drugs including ciclosporin, glibenclamide, and bosentan.
Inhibition of this pump may increase the risk of cholestasis, and the manufacturer of bosentan says that they should be avoided in patients taking bosentan (see ‘glibenclamide’, (p.515) and ‘ciclosporin’, (p.1026)).
1. Lee W, Kim RB. Transporters and renal drug elimination. Annu Rev Pharmacol Toxicol (2004) 44, 137–66.
2. Faber KN, Müller M, Jansen PLM. Drug transport proteins in the liver. Adv Drug Deliv Rev (2003) 55, 107–24.
1.5. Drug transporter proteins
Drugs and endogenous substances are known to cross biological membranes, not just by passive diffusion, but by carrier-mediated processes, often known as transporters. Significant advances in the identification of various transporters have been made, although the contribution of many of these to drug interactions in particular, is still unclear.1,2 The most well known is P-glycoprotein, which is a product of the MDR1 gene (ABCB1 gene) and a member of the ATP-binding cassette (ABC) family of efflux transporters.1 Its involvement in drug interactions is discussed in (a) below.
Another ABC transporter is sister P-glycoprotein, otherwise called the bile salt export pump (BSEP or ABCB11).1 It has been suggested that inhibition of this pump may increase the risk of cholestasis, see Drug transporters under ‘Drug excretion interactions’, (p.7).
Other transporters that are involved in some drug interactions are the organic anion transporters (OATs), organic anion-transporting polypeptides (OATPs) and organic cation transporters (OCTs), which are members of the solute carrier superfamily (SLC) of transporters.1 The best known example
of an OAT inhibitor is probenecid, which affects the renal excretion of a number of drugs, see Changes in active kidney tubule excretion under ‘Drug excretion interactions’, (p.7).

(a) P-glycoprotein interactions
More and more evidence is accumulating to show that some drug interactions occur because they interfere with the activity of P-glycoprotein. This is an efflux pump found in the membranes of certain cells, which can push metabolites and drugs out of the cells and have an impact on the extent of drug absorption (via the intestine), distribution (to the brain, testis, or placenta) and elimination (in the urine and bile). So, for example, the P-glycoprotein in the cells of the gut lining can eject some already-absorbed drug molecules back into the intestine resulting in a reduction in the total amount of drug absorbed. In this way P-glycoprotein acts as a barrier to absorption. The activity of P-glycoprotein in the endothelial cells of the blood-brain barrier can also eject certain drugs from the brain, limiting CNS penetration and effects.
The pumping actions of P-glycoprotein can be induced or inhibited by some drugs. So for example, the induction (or stimulation) of the activity of P-glycoprotein by rifampicin (rifampin) within the lining cells of the gut causes digoxin to be ejected into the gut more vigorously. This results in a fall in the plasma levels of digoxin (see ‘Digitalis glycosides + Rifamycins’, p.938). In contrast, verapamil appears to inhibit the activity of P-glycoprotein, and is well known to increase digoxin levels (see ‘Digitalis glycosides + Calcium-channel blockers; Verapamil’, p.916). Ketoconazole also has P-glycoprotein inhibitory effects, and has been shown to increase CSF levels of ritonavir, possibly by preventing the efflux of ritonavir from the CNS (see ‘Protease inhibitors + Azoles; Ketoconazole’, p.814). Thus the induction or inhibition of P-glycoprotein can have an impact on the pharmacokinetics of some drugs. Note that there is evidence that P-glycoprotein inhibition may have a greater impact on drug distribution (e.g. into the brain) than on drug absorption (e.g. plasma levels). 2 There is an overlap between CYP3A4 and P-glycoprotein inhibitors, inducers and substrates. Therefore, both mechanisms may be involved in many of the drug interactions traditionally thought to be due to changes in CYP3A4. ‘Table 1.6’, (p.8) lists some possible P-glycoprotein inhibitors and inducers. Many drugs that are substrates for CYP3A4 (see ‘Table 1.4’, (p.6)) are also substrates for P-glycoprotein. Digoxin and talinolol are examples of the few drugs that are substrates for P-glycoprotein but not CYP3A4.
P-glycoprotein is also expressed in some cancer cells (where it was first identified). This has led to the development of specific P-glycoprotein inhibitors, such as valspodar, with the aim of improving the penetration of cytotoxic drugs into cancer cells.
1. Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery
and development. Pharmacol Rev (2003) 55, 425–61.
2. Lin JH, Yamazaki M. Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev
(2003) 35, 417–54.
2. Pharmacodynamic interactions
Pharmacodynamic interactions are those where the effects of one drug are changed by the presence of another drug at its site of action. Sometimes the drugs directly compete for particular receptors (e.g. beta2 agonists, such as salbutamol, and beta blockers, such as propranolol) but often the reaction is more indirect and involves interference with physiological mechanisms. These interactions are much less easy to classify neatly than those of a pharmacokinetic type.
2.1. Additive or synergistic interactions
If two drugs that have the same pharmacological effect are given together the effects can be additive. For example, alcohol depresses the CNS and, if taken in moderate amounts with normal therapeutic doses of any of a large number of drugs (e.g. anxiolytics, hypnotics, etc.), may cause excessive drowsiness. Strictly speaking (as pointed out earlier) these are not interactions within the definition given in ‘What is a drug interaction?’, (p.1). Nevertheless, it is convenient to consider them within the broad context of the clinical outcome of giving two drugs together.
Additive effects can occur with both the main effects of the drugs as well as their adverse effects, thus an additive ‘interaction’ can occur with antimuscarinic antiparkinson drugs (main effect) or butyrophenones (adverse effect) that can result in serious antimuscarinic toxicity (see ‘Antipsychotics + Antimuscarinics’, p.708).
Sometimes the additive effects are solely toxic (e.g. additive ototoxicity, nephrotoxicity, bone marrow depression, QT interval prolongation). Examples of these reactions are listed in ‘Table 1.7’, (see below). It is common to use the terms ‘additive’, ‘summation’, ‘synergy’ or ‘potentiation’ to describe what happens if two or more drugs behave like this. These words have precise pharmacological definitions but they are often used rather loosely as synonyms because in practice it is often very difficult to know the extent of the increased activity, that is to say whether the effects are greater or smaller than the sum of the individual effects.
The serotonin syndrome
In the 1950s a serious and life-threatening toxic reaction was reported in patients taking iproniazid (an MAOI) when they were given ‘pethidine (meperidine)’, (p.1140). The reasons were then not understood and even now we do not have the full picture. What happened is thought to have been due to over-stimulation of the 5-HT1A and 5-HT2A receptors and possibly other serotonin receptors in the central nervous system (in the brain stem and spinal cord in particular) due to the combined effects of these two drugs. It can occur exceptionally after taking only one drug, which causes over-stimulation of these 5-HT receptors, but much more usually it develops when two or more drugs (so-called serotonergic or serotomimetic drugs) act in concert. The characteristic symptoms (now known as the serotonin syndrome) fall into three main areas, namely altered mental status (agitation, confusion, mania), autonomic dysfunction (diaphoresis, diarrhoea, fever, shivering) and neuromuscular abnormalities (hyperreflexia, incoordination, myoclonus, tremor). These are the ‘Sternbach diagnostic criteria’ named after Dr Harvey Sternbach who drew up this list of clinical features and who suggested that at least three of them need to be seen before classifying this toxic reaction as the serotonin syndrome rather than the neuroleptic malignant syndrome.1
The syndrome can develop shortly after one serotonergic drug is added to another, or even if one is replaced by another without allowing a long enough washout period in between, and the problem usually resolves within about 24 hours if both drugs are withdrawn and supportive measures given. Non-specific serotonin antagonists (cyproheptadine, chlorpromazine, methysergide) have also been used for treatment. Most patients recover uneventfully, but there have been a few fatalities.

Following the first report of this syndrome, many other cases have been described involving ‘tryptophan and MAOIs’, (p.1151), the ‘tricyclic antidepressants and MAOIs’, (p.1149), and, more recently, the ‘SSRIs’, (p.1142) but other serotonergic drugs have also been involved and the list
continues to grow.
It is still not at all clear why many patients can take two, or sometimes several serotonergic drugs together without problems, while a very small number develop this serious toxic reaction, but it certainly suggests that there are as yet other factors involved that have yet to be identified. The full story is likely to be much more complex than just the simple additive effects of two drugs.
1. Sternbach H. The serotonin syndrome. Am J Psychiatry (1991) 148, 705–13.
2.2. Antagonistic or opposing interactions
In contrast to additive interactions, there are some pairs of drugs with activities that are opposed to one another. For example the coumarins can prolong the blood clotting time by competitively inhibiting the effects of dietary vitamin K. If the intake of vitamin K is increased, the effects of the oral anticoagulant are opposed and the prothrombin time can return to normal, thereby cancelling out the therapeutic benefits of anticoagulant treatment (see ‘Coumarins and related drugs + Vitamin K substances’, p.458).
Other examples of this type of interaction are listed in ‘Table 1.8’, (see below).

2.3. Drug or neurotransmitter uptake interactions
A number of drugs with actions that occur at adrenergic neurones can be prevented from reaching those sites of action by the presence of other drugs. The tricyclic antidepressants prevent the reuptake of noradrenaline (norepinephrine) into peripheral adrenergic neurones. Thus patients taking tricyclics and given parenteral noradrenaline have a markedly increased response (hypertension, tachycardia); see ‘Tricyclic antidepressants + Inotropes and Vasopressors’, p.1237. Similarly, the uptake of guanethidine (and related drugs guanoclor, betanidine, debrisoquine, etc.) is blocked by ‘chlorpromazine, haloperidol, tiotixene’, (p.887), a number of ‘amfetamine-like drugs’, (p.886) and the ‘tricyclic antidepressants’, (p.888) so that the antihypertensive effect is prevented. The antihypertensive effects of clonidine are also prevented by the tricyclic antidepressants, one possible reason being that the uptake of clonidine within the CNS is blocked (see ‘Clonidine + Tricyclic and related antidepressants’, p.884). Some of these interactions at adrenergic neurones are illustrated in ‘Figure 1.5’, (see below).

Fig. 1.5 Interactions at adrenergic neurones. A highly simplified composite diagram of an adrenergic neurone (molecules of noradrenaline (norepinephrine) indicated as (•) contained in a single vesicle at the nerve-ending) to illustrate in outline some of the different sites where drugs can interact. More details of these interactions are to be found in individual monographs.
(''The MAOIs inactivate monoamine oxidase and cause the accumulation of noradrenaline at the nerve ending.
When released by indirectly-acting sympathomimetics this results in a massive stimulation of the receptors and a grossly exaggerated pressor response")
("The tricyclic antidepressants, chlorpromazine, haloperidol, tiotixene, mazindol (?) and pizotifen (?) prevent the uptake of guanethidine and related drugs into the neurones, thereby blocking the antihypertensive effects")
("The tricyclic antidepressants block the uptake mechanism by which noradrenaline is taken into the neurone and removed from the receptor area. As a result the effects of administered noradrenaline are exaggerated")
("Alpha and beta blockers occupy the receptors and prevent the normal stimulant activity of the noradrenaline. Alpha blockers such as phentolamine will block the pressor effects of noradrenaline. Propranolol and similar nonselective beta blockers will antagonise the bronchodilator effects of betaagonist bronchodilators (e.g. salbutamol) ")
("Indirectly-acting sympathomimetics stimulate the release of noradrenaline")
("Directly-acting sympathomimetics act like noradrenaline by direct stimulation of the receptors")
("Mixed action sympathomimetics have both direct and indirect activity")
("Adrenergic nerve ending")
E. Drug-herb interactions
The market for herbal medicines and supplements in the Western world has markedly increased in recent years, and, not surprisingly, reports of interactions with ‘conventional’ drugs have arisen. The most well known and documented example is the interaction of St John’s wort (Hypericum perforatum) with a variety of drugs, see below. There have also been isolated reports of other herbal drug interactions, attributable to various mechanisms, including additive pharmacological effects.
Based on these reports, there are a growing number of reviews of herbal medicine interactions, which seek to predict likely interactions based on the, often hypothesised, actions of various herbs. Many of these predictions seem tenuous at best.
Rather than add to the volume of predicted interactions, at present, Stockley’s Drug Interactions includes only those interactions for which there are published reports.
To aid collection of data in this area, health professionals should routinely ask patients about their use of herbal medicines and supplements, and report any unexpected responses to treatment.
An additional problem in interpreting these interactions, is that the interacting constituent of the herb is usually not known and is therefore not standardised for. It could vary widely between different products, and batches of the same product.
St John’s wort
An increasing number of reports have implicated St John’s wort (Hypericum perforatum) in drug interactions. Evidence has shown that the herb can induce the cytochrome P450 isoenzyme CYP3A4, and can also induce ‘P-glycoprotein’, (p.8). Hence St John’s wort decreases the levels of ‘ciclosporin’, (p.1037) and ‘digoxin’, (p.927), respectively. Other less certain evidence suggests that CYP2E1 and CYP1A2 may also be induced.
St John’s wort has serotonergic properties, and this has resulted in a pharmacodynamic interaction with the ‘SSRIs’, (p.1224), namely the development of the serotonin syndrome. St John’s wort contains many possible constituents that could be responsible for its pharmacological effects. The major active constituents are currently considered to be hyperforin (a phloroglucinol) and hypericin (a naphthodianthrone). Hypericin is the only constituent that is standardised for, and then only in some St John’s wort preparations.
General references
1. Miller LG. Herbal medicinals. Selected clinical considerations focusing on known or potential drug-herb interactions. Arch Intern Med (1998) 158, 2200–11.
2. Fugh-Berman A. Herb-drug interactions. Lancet (2000) 355, 134–8. Correction. ibid. 1020.
3. Wang Z, Gorski JC, Hamman MA, Huang S-M, Lesko LJ, Hall SD. The effects of St John’s wort (Hypericum perforatum) on human cytochrome P450 activity. Clin Pharmacol Ther (2001) 70, 317–26.
4. Williamson EM. Drug interactions between herbal and prescription medicines. Drug Safety (2003) 26, 1075–92.
5. Henderson L, Yue QY, Bergquist C, Gerden B, Arlett P. St John’s wort (Hypericum perforatum): drug interactions and clinical outcomes. Br J Clin Pharmacol (2002) 54, 349–56.
6. Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Cui Y, Ang CYW. Cytochrome P450 phenotypic ratios for predicting herb-drug interactions in humans. Clin Pharmacol Ther (2002) 72, 276–87.
7. Dresser GK, Schwarz UI, Wilkinson GR, Kim RB. Coordinate induction of both cytochrome P4503A and MDR1 by St John’s wort in healthy subjects. Clin Pharmacol Ther (2003) 73, 41–50
F. Drug-food interactionsa
It is well established that food can cause clinically important changes in drug absorption through effects on gastrointestinal motility or by drug binding, see ‘Drug absorption interactions’, (p.3). In addition, it is well known that tyramine (present in some foodstuffs) may reach toxic concentrations in patients taking ‘MAOIs’, (p.1153). With the growth in understanding of drug metabolism mechanisms, it has been increasingly recognised that some foods can alter drug metabolism. Currently, grapefruit juice causes the most clinically relevant of these interactions, see (b) below.
(a) Cruciferous vegetables and charcoal-broiled meats
Cruciferous vegetables, such as brussels sprouts, cabbage, and broccoli, contain substances that are inducers of the cytochrome P450 isoenzyme CYP1A2. Chemicals formed by ‘burning’ meats additionally have these properties. These foods do not appear to cause any clinically important drug interactions in their own right, but their consumption may add another variable to drug interaction studies, so complicating interpretation. In drug interaction studies where alteration of CYP1A2 is a predicted mechanism, it may be better for patients to avoid these foods during the study.
(b) Grapefruit juice
By chance, grapefruit juice was chosen to mask the taste of alcohol in a study of the effect of alcohol on felodipine, which led to the discovery that grapefruit juice itself markedly increased felodipine levels, see ‘Calciumchannel blockers + Grapefruit juice’, p.869. In general, grapefruit juice inhibits intestinal CYP3A4, and only slightly affects hepatic CYP3A4. This is demonstrated by the fact that intravenous preparations of drugs that are metabolised by CYP3A4 are not much affected, whereas oral preparations of the same drugs are. These interactions result in increased drug levels.
Some drugs that are not metabolised by CYP3A4 show decreased levels with grapefruit juice, such as ‘fexofenadine’, (p.588). The probable reason for this is that grapefruit juice is an inhibitor of some drug transporters (see ‘Drug transporter proteins’, (p.8)), and possibly affects organic aniontransporting polypeptides (OATPs), although inhibition of P-glycoprotein has also been suggested.
The active constituent of grapefruit juice is uncertain. Grapefruit contains naringin, which degrades during processing to naringenin, a substance known to inhibit CYP3A4. Because of this, it has been assumed that whole grapefruit will not interact, but that processed grapefruit juice will. However, subsequently some reports have implicated the whole fruit. Other possible active constituents in the whole fruit include bergamottin and dihydroxybergamottin.
General references
1. Ameer B, Wientraub RA. Drug interactions with grapefruit juice. Clin Pharmacokinet (1997) 33, 103–21.
G. Conclusions
It is now quite impossible to remember all the known clinically important interactions and how they occur, which is why this reference publication has been produced, but there are some broad general principles that need little memorising:
• Be on the alert with any drugs that have a narrow therapeutic window or where it is necessary to keep serum levels at or above a suitable level (e.g. anticoagulants, antidiabetic drugs, antiepileptics, antihypertensives, anti-infectives, antineoplastic cytotoxics, digitalis glycosides, immunosuppressants, etc.).
• Remember some of those drugs that are key enzyme inducers (e.g. phenytoin, barbiturates, rifampicin, etc) or enzyme inhibitors (e.g. azole antifungals, HIV-protease inhibitors, erythromycin, SSRIs).
• Think about the basic pharmacology of the drugs under consideration so that obvious problems (additive CNS depression for example) are not overlooked, and try to think what might happen if drugs that affect the same receptors are used together. And don’t forget that many drugs affect more than one type of receptor.
• Keep in mind that the elderly are at risk because of reduced liver and renal function on which drug clearance depends.
2
ACE inhibitors and Angiotensin II receptor antagonists
ACE inhibitors (angiotensin-converting enzyme inhibitors) prevent the production of angiotensin II from angiotensin I. The angiotensin II receptor antagonists are more selective, and target the angiotensin II type I (AT1) receptor, which is responsible for the pressor actions of angiotensin II.
Angiotensin II is involved in the renin-angiotensin-aldosterone system, which regulates blood pressure, sodium and water homoeostasis by the kidneys, and cardiovascular function. Angiotensin II stimulates the synthesis and secretion of aldosterone and raises blood pressure via a direct vasoconstrictor effect.
Angiotensin converting enzyme (ACE) is identical to bradykinase, so ACE inhibitors may additionally reduce the degradation of bradykinin and affect enzymes involved in the production of prostaglandins.
Many of the interactions of the ACE inhibitors and angiotensin II receptor antagonists involve drugs that affect blood pressure. Consequently in most cases the result is either an increase in the hypotensive effect (e.g. ‘alcohol’, (p.48)) or a decrease in the hypotensive effect (e.g. ‘indometacin’, (p.28)).
In addition, due to their effects on aldosterone, the ACE inhibitors and angiotensin II antagonists may increase potassium concentrations and can therefore have additive hyperkalaemic effects with other drugs that cause elevated potassium levels. Furthermore, drugs that affect renal function may potentiate the adverse effects of ACE inhibitors and angiotensin II antagonists on the kidneys.
Most ACE inhibitor and angiotensin II receptor antagonist interactions are pharmacodynamic, that is, interactions that result in an alteration in drug effects rather than drug disposition, so in most cases interactions of individual drugs will be applicable to the group. In vitro experiments suggest that the role of cytochrome P450 isoenzymes in the metabolism and interactions of the angiotensin II receptor antagonists (candesartan, eprosartan, irbesartan, losartan and valsartan) is small, although losartan, irbesartan, and to a minor extent, candesartan, are metabolised by CYP2C9.
Only losartan and irbesartan were considered to have a theoretical potential for pharmacokinetic drug interactions involving the CYP2C9 enzyme. 1 See ‘Angiotensin II receptor antagonists + Azoles’, p.35. The ACE inhibitors do not appear to undergo interactions via cytochrome P450 isoenzymes.
‘Table 2.1’, (see below) lists the ACE inhibitors and the angiotensin II receptor antagonists. Although most of the interactions of the ACE inhibitors or angiotensin II receptor antagonists are covered in this section, if the ACE inhibitor or angiotensin II receptor antagonist is the affecting drug, the interaction is dealt with elsewhere.
1. Taavitsainen P, Kiukaanniemi K, Pelkonen O. In vitro inhibition screening of human hepatic P450 enzymes by five angiotensin-II receptor antagonists. Eur J Clin Pharmacol (2000) 56, 135–40.

Angiotensin II is involved in the renin-angiotensin-aldosterone system, which regulates blood pressure, sodium and water homoeostasis by the kidneys, and cardiovascular function. Angiotensin II stimulates the synthesis and secretion of aldosterone and raises blood pressure via a direct vasoconstrictor effect.
Angiotensin converting enzyme (ACE) is identical to bradykinase, so ACE inhibitors may additionally reduce the degradation of bradykinin and affect enzymes involved in the production of prostaglandins.
Many of the interactions of the ACE inhibitors and angiotensin II receptor antagonists involve drugs that affect blood pressure. Consequently in most cases the result is either an increase in the hypotensive effect (e.g. ‘alcohol’, (p.48)) or a decrease in the hypotensive effect (e.g. ‘indometacin’, (p.28)).
In addition, due to their effects on aldosterone, the ACE inhibitors and angiotensin II antagonists may increase potassium concentrations and can therefore have additive hyperkalaemic effects with other drugs that cause elevated potassium levels. Furthermore, drugs that affect renal function may potentiate the adverse effects of ACE inhibitors and angiotensin II antagonists on the kidneys.
Most ACE inhibitor and angiotensin II receptor antagonist interactions are pharmacodynamic, that is, interactions that result in an alteration in drug effects rather than drug disposition, so in most cases interactions of individual drugs will be applicable to the group. In vitro experiments suggest that the role of cytochrome P450 isoenzymes in the metabolism and interactions of the angiotensin II receptor antagonists (candesartan, eprosartan, irbesartan, losartan and valsartan) is small, although losartan, irbesartan, and to a minor extent, candesartan, are metabolised by CYP2C9.
Only losartan and irbesartan were considered to have a theoretical potential for pharmacokinetic drug interactions involving the CYP2C9 enzyme. 1 See ‘Angiotensin II receptor antagonists + Azoles’, p.35. The ACE inhibitors do not appear to undergo interactions via cytochrome P450 isoenzymes.
‘Table 2.1’, (see below) lists the ACE inhibitors and the angiotensin II receptor antagonists. Although most of the interactions of the ACE inhibitors or angiotensin II receptor antagonists are covered in this section, if the ACE inhibitor or angiotensin II receptor antagonist is the affecting drug, the interaction is dealt with elsewhere.
1. Taavitsainen P, Kiukaanniemi K, Pelkonen O. In vitro inhibition screening of human hepatic P450 enzymes by five angiotensin-II receptor antagonists. Eur J Clin Pharmacol (2000) 56, 135–40.

ACE inhibitors + Allopurinol
Three cases of Stevens-Johnson syndrome (one fatal) and two cases of hypersensitivity have been attributed to the use of captopril with allopurinol. Anaphylaxis and myocardial infarction occurred in one man taking enalapril when given allopurinol. The combination of ACE inhibitors and allopurinol may increase the risk of leucopenia and serious infection, especially in renal impairment.
Clinical evidence
An elderly man with hypertension, chronic renal failure, congestive heart failure and mild polyarthritis receiving multiple drug treatment, which included captopril 25 mg twice daily and diuretics, developed fatal Stevens-Johnson syndrome about 5 weeks after starting to take allopurinol 100 mg twice daily.1 The authors of the report noted that the manufacturer of captopril was aware of two other patients who developed the syndrome 3 to 5 weeks after allopurinol was started.1 Another report describes fever, arthralgia and myalgia in a diabetic man with chronic renal failure who was also given captopril and allopurinol. He improved when the captopril was withdrawn.2 Exfoliatory facial dermatitis occurred in a patient with renal failure who was taking captopril and allopurinol.3 A man taking enalapril had an acute anaphylactic reaction with severe coronary spasm, culminating in myocardial infarction, within 20 minutes of taking allopurinol 100 mg. He recovered and continued to take enalapril without allopurinol.4
The UK manufacturer of captopril also warns that neutropenia and agranulocytosis, resulting in serious infection, have occurred in patients taking captopril and other ACE inhibitors, and that concurrent treatment with allopurinol may be a complicating factor, especially in those with renal impairment.5 However, the US manufacturer notes that, while renal impairment and a relatively high dose of captopril markedly increases the risk of neutropenia, no association between allopurinol and captopril and neutropenia has appeared in US reports.6
No significant pharmacokinetic changes were seen in 12 healthy subjects given allopurinol and captopril alone and in combination.7
Mechanism
Not understood. It is uncertain whether these are interactions because allopurinol alone can cause severe hypersensitivity reactions, particularly in the presence of renal failure and in conjunction with diuretic use. Captopril can also induce a hypersensitivity reaction.
Importance and management
These interactions are not clearly established, and the reaction appears to be rare and unpredictable. All that can be constructively said is that patients taking both drugs should be very closely monitored for any signs of hypersensitivity (e.g. skin reactions) or low white cell count (sore throat, fever), especially if they have renal impairment. The UK manufacturer of captopril recommends that differential white blood cell counts should be performed before adding allopurinol, then every 2 weeks during the first 3 months of treatment, and periodically thereafter.5 Similar caution and advice is given by the UK manufacturers of several other ACE inhibitors.
For other possible interactions with ACE inhibitors that might result in an increased risk of leucopenia see also ‘ACE inhibitors + Azathioprine’, p.18 and ‘ACE inhibitors + Procainamide’, p.33.
1. Pennell DJ, Nunan TO, O’Doherty MJ, Croft DN. Fatal Stevens-Johnson syndrome in a patient on captopril and allopurinol. Lancet (1984) i, 463.
2. Samanta A, Burden AC. Fever, myalgia, and arthralgia in a patient on captopril and allopurinol. Lancet (1984) i, 679.
3. Beeley L, Daly M, Stewart P. Bulletin of the West Midlands Centre for Adverse Drug Reaction Reporting (1987) 24, 9.
4. Ahmad S. Allopurinol and enalapril. Drug induced anaphylactic coronary spasm and acute myocardial infarction. Chest (1995) 108, 586.
5. Capoten (Captopril). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Capoten (Captopril). Par Pharmaceutical, Inc. US Prescribing information, June 2003.
7. Duchin KL, McKinstry DN, Cohen AI, Migdalof BH. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin Pharmacokinet (1988) 14, 241–59.
ACE inhibitors + Angiotensin II receptor antagonists
The combined use of ACE inhibitors and angiotensin II receptor antagonists increases the risk of hypotension, renal impairment and hyperkalaemia in patients with heart failure.
Clinical evidence, mechanism, importance and management
Both ACE inhibitors and angiotensin II receptor antagonists can have adverse renal effects and can cause hyperkalaemia. These effects might be expected to be additive when they are used together. In one randomised clinical study in patients with heart failure taking ACE inhibitors, the addition of candesartan resulted in higher rates of withdrawals than placebo for renal impairment (increase in creatinine 7.8% versus 4.1%) and hyperkalaemia (3.4% versus 0.7%).1 In another double-blind study in patients with heart failure, the combination of valsartan and captopril resulted in a higher incidence of adverse events leading to a dose reduction or a discontinuation of study treatment than either drug alone. For hypotension, treatment was discontinued in 90 (1.9%) of patients in the combined group, 70 (1.4%) of patients in the valsartan group, and 41 (0.8%) of patients in the captopril group. For renal causes the corresponding figures were 61 (1.3%), 53 (1.1%) and 40 (0.8%) of patients, respectively, and for hyperkalaemia the figures were 12 (0.2%), 7 (0.1%) and 4 (0.1%) of patients, respectively.2
Monitor renal function and serum potassium carefully when combination therapy is used.
1. McMurray JJV, Östergren J, Swedberg K, Granger CB, Held P, Michelson EL, Olofsson B, Yusuf S, Pfeffer MA; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin- converting-enzyme inhibitors: the CHARM-Added trial. Lancet (2003) 362, 767–71.
2. Pfeffer MA, McMurray JJV, Velazquez EJ, Rouleau J-L, Køber L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM; Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med (2003) 349, 1893–1906.
ACE inhibitors + Antacids
An aluminium/magnesium hydroxide antacid reduced the bioavailability of captopril by 40%, but this did not seem to be clinically important. The bioavailability of fosinopril was reduced by about one-third by Mylanta. An antacid did not affect ramipril pharmacokinetics.
Clinical evidence
In 10 healthy subjects an antacid containing aluminium/magnesium hydroxide and magnesium carbonate reduced the AUC of a single 50-mg dose of captopril by about 40%, when compared with the fasting state.
However, this did not alter the extent of the reduction in blood pressure.1
Another study found that Mylanta [aluminium/magnesium hydroxide and simeticone2] reduced the bioavailability of fosinopril 20 mg by about one-third.3
It is briefly noted in a review that antacid use did not affect the pharmacokinetics of ramiprilat, the active metabolite of ramipril.4
Mechanism
The mechanism of this interaction is uncertain, but is unlikely to be due to elevated gastric pH since cimetidine did not have a similar effect.3
Importance and management
Note that greater decreases in captopril bioavailability (caused by ‘food’, (p.26)) were found not to be clinically relevant, therefore, it is unlikely the change seen with antacids will be clinically important.
However, with fosinopril, the manufacturers2,5 suggest separating administration of antacids by at least 2 hours.
The UK manufacturers of quinapril6 and trandolapril7 also warn that antacids may reduce the bioavailability of ACE inhibitors, quite possibly based on the way these named ACE inhibitors interact, but there seems to be no evidence of a clinically significant interaction in practice.
1. Mäntylä R, Männistö PT, Vuorela A, Sundberg S, Ottoila P. Impairment of captopril bioavailability by concomitant food and antacid intake. Int J Clin Pharmacol Ther Toxicol (1984) 22, 626–9.
2. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
3. Moore L, Kramer A, Swites B, Kramer P, Tu J. Effect of cimetidine and antacid on the kinetics of the active diacid of fosinopril in healthy subjects. J Clin Pharmacol (1988) 28, 946.
4. Todd PA, Benfield P. Ramipril. A review of its pharmacological properties and therapeutic efficacy in cardiovascular disorders. Drugs (1990) 39, 110–35.
5. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Accupro (Quinapril hydrochloride). Pfizer Ltd. UK Summary of product characteristics, March 2007.
7. Gopten (Trandolapril). Abbott Laboratories Ltd. UK Summary of product characteristics, May 2007.
ACE inhibitors + Antipsychotics
Marked postural hypotension occurred in a patient given chlorpromazine and captopril. The hypotensive adverse effects of antipsychotics such as the phenothiazines may be additive with the effects of ACE inhibitors.
Clinical evidence, mechanism, importance and management A patient fainted and developed marked postural hypotension (standing blood pressure 66/48 mmHg) when given captopril 6.25 mg twice daily and chlorpromazine 200 mg three times daily. He had previously taken chlorpromazine with nadolol, prazosin and hydrochlorothiazide without any problems, although his blood pressure was poorly controlled on these drugs. Since the patient’s blood pressure was quite elevated when taking chlorpromazine or captopril alone, there appeared to be a synergistic hypotensive effect between the two drugs.1
The manufacturers of several ACE inhibitors warn that ACE inhibitors may enhance the hypotensive effects of certain antipsychotics, and that postural hypotension may occur. Some of these warnings are based, not unreasonably, on the adverse reactions seen with other ACE inhibitors or antihypertensives, but not necessarily on direct observations.2 If postural hypotension occurs warn patients to lay down and elevate their legs if they feel faint or dizzy, and, when recovered, to get up slowly. Dosage adjustments may be necessary to accommodate this interaction.
1. White WB. Hypotension with postural syncope secondary to the combination of chlorpromazine and captopril. Arch Intern Med (1986) 146, 1833–4.
2. Knoll Ltd. Personal communication,1993.
ACE inhibitors + Aprotinin
Aprotinin suppressed the hypotensive action of captopril and enalapril in rats.
Clinical evidence, mechanism, importance and management
A study in spontaneously hypertensive rats found that aprotinin suppressed the hypotensive responses of captopril and enalapril.1 Aprotinin is a proteolytic enzyme inhibitor that has many actions including antagonism of the kallikrein-kinin system, which in turn affects bradykinins and renin. It would therefore be expected to have complex interactions with the ACE inhibitors,2 which also affect these proteins. However, there does not appear to be any evidence to suggest that this theoretical interaction is of clinical relevance.
1. Sharma JN, Amrah SS, Noor AR. Suppression of hypotensive responses of captopril and enalapril by the kallikrein inhibitor aprotinin in spontaneously hypertensive rats. Pharmacology (1995) 50, 363–9.
2. Waxler B, Rabito SF. Aprotinin: a serine protease inhibitor with therapeutic actions: its interaction with ACE inhibitors. Curr Pharm Des (2003) 9, 777–87.
ACE inhibitors + Aspirin
The antihypertensive efficacy of captopril and enalapril may be reduced by high-dose aspirin in about 50% of patients. Low-dose aspirin (less than or equal to 100 mg daily) appears to have little effect. It is unclear whether aspirin attenuates the benefits of ACE inhibitors in heart failure. The likelihood of an interaction may depend on disease state and its severity.
Renal failure has been reported in a patient taking captopril and aspirin.
Clinical evidence
A. Effects on blood pressure
(a) Captopril
Aspirin 600 mg every 6 hours for 5 doses did not significantly alter the blood pressure response to a single 25 to 100-mg dose of captopril in 8 patients with essential hypertension. However, the prostaglandin response to captopril was blocked in 4 of the 8, and in these patients, the blood pressure
response to captopril was blunted.1 In another study, aspirin 75 mg daily did not alter the antihypertensive effects of captopril 25 mg twice daily in 15 patients with hypertension.2
(b) Enalapril
Two groups of 26 patients, one with mild to moderate hypertension taking enalapril 20 mg twice daily and the other with severe primary hypertension taking enalapril 20 mg twice daily (with nifedipine 30 mg and atenolol 50 mg daily), were given test doses of aspirin 100 and 300 mg daily for 5 days. The 100-mg dose of aspirin did not alter the efficacy of the antihypertensive drugs, but the 300-mg dose reduced the antihypertensive efficacy in about half the patients in both groups. In these patients, the antihypertensive effects were diminished by 63% in those with mild to moderate hypertension and by 91% in those with severe hypertension.3 In contrast, another study in 7 patients with hypertension taking enalapril (mean daily dose 12.9 mg) found that aspirin 81 mg or 325 mg daily for 2 weeks did not have any significant effect on blood pressure.4 A further study in 18 patients also found that aspirin 100 mg daily for 2 weeks did not alter the antihypertensive effect of enalapril 20 or 40 mg daily.5
(c) Unspecified ACE inhibitors
In a randomised study, the use of low-dose aspirin 100 mg daily for 3 months did not alter blood pressure control in patients taking calciumchannel blockers or ACE inhibitors, when compared with placebo.6 Similarly, in a re-analysis of data from the Hypertension Optimal Treatment (HOT) study, long-term low-dose aspirin 75 mg daily did not interfere with the blood pressure-lowering effects of the antihypertensive drugs studied, when compared with placebo. Of 18 790 treated hypertensive patients, about 82% received a calcium-channel blocker, usually felodipine alone or in combination, and 41% received an ACE inhibitor, usually in combination with felodipine.7
B. Effects in coronary artery disease and heart failure
Various pharmacological studies have looked at the short-term effects of the combination of ACE inhibitors and aspirin on haemodynamic parameters.
In one study in 40 patients with decompensated heart failure, aspirin 300 mg given on the first day and 100 mg daily thereafter antagonised the short-term haemodynamic effects of captopril 50 mg given every 8 hours for 4 days. The captopril-induced increase in cardiac index and the reduction in peripheral vascular resistance and pulmonary wedge pressure were all abolished.8 In another study, in 15 patients with chronic heart failure receiving treatment with ACE inhibitors (mainly enalapril 10 mg twice daily), aspirin in doses as low as 75 mg impaired vasodilatation induced by arachidonic acid.9 In yet another study, aspirin 325 mg daily worsened pulmonary diffusion capacity and made the ventilatory response to exercise less effective in patients taking enalapril 10 mg twice daily, but did not exert this effect in the absence of ACE inhibitors.10 However, results from studies are inconsistent. In a review,11 five of 7 studies reported aspirin did not alter the haemodynamic effects of ACE inhibitors whereas the remaining two did. In one of these studies showing an adverse interaction between aspirin and enalapril, ticlopidine did not interact with enalapril.12
A number of large clinical studies of ACE inhibitors, mostly post-myocardial infarction, have been re-examined to see if there was a difference in outcome between those receiving aspirin at baseline, and those not. The results are summarised in ‘Table 2.2’, (p.15). However, in addition to the problems of retrospective analysis of non-randomised parameters, the studies vary in the initiation and duration of aspirin and ACE inhibitor treatment and the length of follow-up, the degree of heart failure or ischaemia, the prognosis of the patients, and the final end point (whether compared with placebo or with the benefits of aspirin or ACE inhibitors). The conclusions are therefore conflicting, and, although two meta-analyses of these studies found no interaction, an editorial13 disputes the findings of one of these analyses.14 In addition to these sub-group analyses, there have been a number of retrospective cohort studies. A retrospective study involving 576 patients with heart failure requiring hospitalisation, showed a trend towards an increased incidence of early readmissions (within 30 days after discharge) for heart failure among subjects treated with ACE inhibitors and aspirin, compared with those treated with ACE inhibitors without aspirin (16% versus 10%). In patients without coronary artery disease the increase in readmissions was statistically significant (23% versus 10%).15 However, long-term survival in heart failure was not affected by the use of aspirin with ACE inhibitors. Furthermore, among patients with coronary artery disease there was a trend towards improvement in mortality in patients treated with the combination, compared with ACE inhibitor without aspirin (40% versus 56%).16 Similarly, a lack of adverse interaction was found in a retrospective study involving 14 129 elderly patients who survived a hospitalisation for acute myocardial infarction. However, the added benefit of the combination over patients who received either aspirin or ACE inhibitors alone was not statistically significant.17 Similarly, in another cohort of patients discharged after first hospitalisation for heart failure, there was no increase in mortality rates or readmission rates in those taking aspirin and ACE inhibitors.18 In another retrospective analysis in patients with stable left ventricular systolic dysfunction, no decrease in survival was seen in patients receiving ACE inhibitors, when comparing those also receiving aspirin (mean dose 183 mg daily, 74% 200 mg or less) and those not.19 Conversely, another study found that, compared to patients not taking aspirin, the use of high-dose aspirin (325 mg daily or more) with an ACE inhibitor was associated with a small but statistically significant 3% increase in the risk of death, whereas low-dose aspirin (160 mg daily or less) was not.20
C. Effects on renal function
Acute renal failure developed in a woman taking captopril when she started to take aspirin for arthritis. Renal function improved when both were stopped.21 However, in a re-analysis of data from the Hypertension Optimal Treatment (HOT) study, long-term low-dose aspirin 75 mg daily had no effect on changes in serum creatinine, estimated creatinine clearance or the number of patients developing renal impairment, when compared with placebo. Of 18 790 treated hypertensive patients, 41% received an ACE inhibitor.7
D. Pharmacokinetic studies
A single-dose study in 12 healthy subjects found that the pharmacokinetics of benazepril 20 mg and aspirin 325 mg were not affected by concurrent use.22
Mechanism
Some, but not all the evidence suggests that prostaglandins may be involved in the hypotensive action of ACE inhibitors, and that aspirin, by inhibiting prostaglandin synthesis, may partially antagonise the effect of ACE inhibitors on blood pressure. This effect appears to depend on the dose of aspirin and may also be dependent on sodium status and plasma renin, and therefore it does not occur in all patients.
The beneficial effects of ACE inhibitors in heart failure and ischaemic heart disease are thought to be due, in part, to the inhibition of the breakdown of kinins, which are important regulators of prostaglandin and nitric oxide synthesis. Such inhibition promotes vasodilatation and afterload reduction.
Aspirin may block these beneficial effects by inhibiting cyclo-oxygenase (COX) and thus prostaglandin synthesis, causing vasoconstriction, decreased cardiac output and worsening heart failure.11,23
Importance and management
Low-dose aspirin (less than or equal to 100 mg daily) does not alter the antihypertensive efficacy of captopril and enalapril. No special precautions would therefore seem to be required with ACE inhibitors and these low doses of aspirin. A high dose of aspirin (2.4 g daily) has been reported to interact in 50% of patients in a single study. Aspirin 300 mg daily has been reported to interact in about 50% of patients in another study, whereas 325 mg daily did not interact in further study. Thus, at present, it appears that if an ACE inhibitor is used with aspirin in doses higher than 300 mg daily, blood pressure should be monitored more closely, and the ACE inhibitor dosage raised if necessary. Intermittent use of aspirin should be considered as a possible cause of erratic control of blood pressure in patients on ACE inhibitors.
Both ACE inhibitors and aspirin are often taken by patients with coronary artery disease, and ACE inhibitors are used in chronic heart failure, which is often associated with coronary heart disease. The
information about a possible interaction between ACE inhibitors and aspirin in heart failure is conflicting. This may be due to much of the clinical data being obtained from retrospective non-randomised analyses.23 It may also be a factor of different disease states. For example, an interaction may be less likely to be experienced in patients with heart failure of ischaemic aetiology than those with non-ischaemic causes, because of the added benefits of aspirin in ischaemic heart disease.24 The available data, and its implications, have been extensively reviewed and commented on.11,13,23-32 Some commentators have advised that, if possible, aspirin should be avoided in patients requiring long-term treatment for heart failure, particularly if heart failure is severe.13,27 Others suggest avoiding aspirin in heart failure unless there are clear indications, such as atherosclerosis.
11,23,28,29 The use of lower doses of aspirin (80 to 100 mg daily rather than greater than or equal to 325 mg daily) in those with heart failure taking ACE inhibitors has also been suggested.24,25,28 US guidelines from 2005 on chronic heart failure33 state that, “Many physicians believe the data justify prescribing aspirin and ACE inhibitors together when there is an indication for use of aspirin,” while recognising that not all physicians agree. The guidelines say that further study is needed. European guidelines state that there is little evidence to support using ACE inhibitors and aspirin together in heart failure. The guidelines say aspirin can be used as prophylaxis after prior myocardial infarction, but that it should be avoided in patients with recurrent hospitalisation for worsening heart failure.34 NICE guidelines in the UK make no comment about the combination of ACE inhibitors and aspirin. They say that all patients with heart failure due to left ventricular systolic dysfunction should be considered for treatment with an ACE inhibitor, and that aspirin (75 to 150 mg once daily) should be prescribed for patients with the combination of heart failure and atherosclerotic arterial disease (including coronary heart disease).35 Data from ongoing randomised studies may provide further insight. Until these are available, combined low-dose aspirin and ACE inhibitors may continue to be used where there is a clear indication for both.
An increased risk of deterioration in renal function or acute renal failure appears to occur rarely with the combination of aspirin and ACE inhibitors. The routine monitoring of renal function, which is advised with ACE inhibitors, should be sufficient to detect any interaction.
1. Moore TJ, Crantz FR, Hollenberg NK, Koletsky RJ, Leboff MS, Swartz SL, Levine L, Podolsky S, Dluhy RG, Williams GH. Contribution of prostaglandins to the antihypertensive action of captopril in essential hypertension. Hypertension (1981) 3, 168–73.
2. Smith SR, Coffman TM, Svetkey LP. Effect of low-dose aspirin on thromboxane production and the antihypertensive effect of captopril. J Am Soc Nephrol (1993) 4, 1133–9.
3. Guazzi MD, Campodonico J, Celeste F, Guazzi M, Santambrogio G, Rossi M, Trabattoni D, Alimento M. Antihypertensive efficacy of angiotensin converting enzyme inhibition and aspirin counteraction. Clin Pharmacol Ther (1998) 63, 79–86.
4. Nawarskas JJ, Townsend RR, Cirigliano MD, Spinler SA. Effect of aspirin on blood pressure in hypertensive patients taking enalapril or losartan. Am J Hypertens (1999) 12, 784–9.
5. Polónia J, Boaventura I, Gama G, Camões I, Bernardo F, Andrade P, Nunes JP, Brandão F, Cerqueira-Gomes M. Influence of non-steroidal anti-inflammatory drugs on renal function and 24 h ambulatory blood pressure-reducing effects of enalapril and nifedipine gastrointestinal therapeutic system in hypertensive patients. J Hypertens (1995) 13, 925–31
6. Avanzini F, Palumbo G, Alli C, Roncaglioni MC, Ronchi E, Cristofari M, Capra A, Rossi S, Nosotti L, Costantini C, Pietrofeso R. Collaborative Group of the Primary Prevention Project (PPP)–Hypertension study. Effects of low-dose aspirin on clinic and ambulatory blood pressure in treated hypertensive patients. Am J Hypertens (2000) 13, 611–16.
7. Zanchetti A, Hansson L, Leonetti G, Rahn K-H, Ruilope L, Warnold I, Wedel H. Low-dose aspirin does not interfere with the blood pressure-lowering effects of antihypertensive therapy. J Hypertens (2002) 20, 1015–22.
8. Viecili PR, Pamplona D, Park M, Silva SR, Ramires JAF, da Luz PL. Antagonism of the acute hemodynamic effects of captopril in decompensated congestive heart failure by aspirin administration. Braz J Med Biol Res (2003) 36, 771–80.
9. Davie AP, Love MP, McMurray JJV. Even low-dose aspirin inhibits arachidonic acid-induced vasodilation in heart failure. Clin Pharmacol Ther (2000) 67, 530–7.
10. Guazzi M, Pontone G, Agostoni P. Aspirin worsens exercise performance and pulmonary gas exchange in patients with heart failure who are taking angiotensin-converting enzyme inhibitors. Am Heart J (1999) 138, 254–60.
11. Mahé I, Meune C, Diemer M, Caulin C, Bergmann J-F. Interaction between aspirin and ACE inhibitors in patients with heart failure. Drug Safety (2001) 24, 167–82.
12. Spaulding C, Charbonnier B, Cohen-Solal A, Juillière Y, Kromer EP, Benhamda K, Cador R, Weber S. Acute hemodynamic interaction of aspirin and ticlopidine with enalapril. Results of a double-blind, randomized comparative trial. Circulation (1998) 98, 757–65.
13. Hall D. The aspirin—angiotensin-converting enzyme inhibitor tradeoff: to halve and halve not. J Am Coll Cardiol (2000) 35, 1808–12.
14. Latini R, Tognoni G, Maggioni AP, Baigent C, Braunwald E, Chen Z-M, Collins R, Flather M, Franzosi MG, Kjekshus J, Køber L, Liu L-S, Peto R, Pfeffer M, Pizzetti F, Santoro E, Sleight P, Swedberg K, Tavazzi L, Wang W, Yusuf S, on behalf of the Angiotensin-converting Enzyme Inhibitor Myocardial Infarction Collaborative Group. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for acute myocardial infarction are similar in the presence and absence of aspirin. Systematic overview of individual data from 96,712 randomized patients. J Am Coll Cardiol (2000) 35 1801–7.
15. Harjai KJ, Nunez E, Turgut T, Newman J. Effect of combined aspirin and angiotensin-converting
enzyme inhibitor therapy versus angiotensin-converting enzyme inhibitor therapy alone on readmission rates in heart failure. Am J Cardiol (2001) 87, 483–7.
16. Harjai KJ, Solis S, Prasad A, Loupe J. Use of aspirin in conjunction with angiotensin-converting
enzyme inhibitors does not worsen long-term survival in heart failure. Int J Cardiol (2003) 88, 207–14.
17. Krumholz HM, Chen Y-T, Wang Y, Radford MJ. Aspirin and angiotensin-converting enzyme inhibitors among elderly survivors of hospitalization for an acute myocardial infarction.
Arch Intern Med (2001) 161, 538–44.
18. McAlister FA, Ghali WA, Gong Y, Fang J, Armstrong PW, Tu JV. Aspirin use and outcomes in a community-based cohort of 7352 patients discharged after first hospitalization for heart failure. Circulation (2006) 113, 2572–8.
19. Aumégeat V, Lamblin N, de Groote P, Mc Fadden EP, Millaire A, Bauters C, Lablanche JM. Aspirin does not adversely affect survival in patients with stable congestive heart failure treated with angiotensin-converting enzyme inhibitors. Chest (2003) 124, 1250–8.
20. Guazzi M, Brambilla R, Rèina G, Tumminello G, Guazzi MD. Aspirin-angiotensin-converting enzyme inhibitor coadministration and mortality in patients with heart failure: a dose-related adverse effect of aspirin. Arch Intern Med (2003) 163, 1574–9.
21. Seelig CB, Maloley PA, Campbell JR. Nephrotoxicity associated with concomitant ACE inhibitor
and NSAID therapy. South Med J (1990) 83, 1144–8.
22. Sioufi A, Pommier F, Gauducheau N, Godbillon J, Choi L, John V. The absence of a pharmacokinetic
interaction between aspirin and the angiotensin-converting enzyme inhibitor
benazepril in healthy volunteers. Biopharm Drug Dispos (1994) 15, 451–61.
23. Massie BM, Teerlink JR. Interaction between aspirin and angiotensin-converting enzyme inhibitors:
real or imagined. Am J Med (2000) 109, 431–3.
24. Nawarskas JJ, Spinler SA. Update on the interaction between aspirin and angiotensin-converting
enzyme inhibitors. Pharmacotherapy (2000) 20, 698–710.
25. Stys T, Lawson WE, Smaldone GC, Stys A. Does aspirin attenuate the beneficial effects of angiotensin-converting enzyme inhibition in heart failure? Arch Intern Med (2000) 160, 1409–13.
26. Barbash IM, Goldbourt U, Gottlieb S, Behar S, Leor J. Possible interaction between aspirin and ACE inhibitors: update on unresolved controversy. Congest Heart Fail (2000) 6, 313–18.
27. Cleland JGF, John J, Houghton T. Does aspirin attenuate the effect of angiotensin-converting enzyme inhibitors in hypertension or heart failure? Curr Opin Nephrol Hypertens (2001) 10, 625–31.
28. Peterson JG, Lauer MS. Using aspirin and ACE inhibitors in combination: why the hullabaloo? Cleve Clin J Med (2001) 68, 569–74.
29. Olson KL. Combined aspirin/ACE inhibitor treatment for CHF. Ann Pharmacother (2001) 35, 1653–8.
30. Park MH. Should aspirin be used with angiotensin-converting enzyme inhibitors in patients with chronic heart failure? Congest Heart Fail (2003) 9, 206–11.
31. Konstam MA. Aspirin and heart failure: square evidence meets a round patient. Congest Heart Fail (2003) 9, 203–5.
32. Brunner-La Rocca HP. Interaction of angiotensin-converting enzyme inhibition and aspirin in congestive heart failure: long controversy finally resolved? Chest (2003) 124, 1192–4.
33. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation (2005) 112, e154–235. Available at: http://circ.ahajournals.org/cgi/content/full/ 112/12/e154 (accessed 14/08/07).
34. Swedberg K, Cleland J, Dargie H, Drexler H, Follath F, Komajda M, Tavazzi L, Smiseth OA, Gavazzi A, Haverich A, Hoes A, Jaarsma T, Korewicki J, Lévy S, Linde C, Lopez-Sendon JL, Nieminen MS, Piérard L, Remme WJ; The Task Force for the Diagnosis and Treatment of CHF of the European Society of Cardiology. Guidelines for the diagnosis and treatment of chronic heart failure: full text (update 2005): The Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology. Eur Heart J (2005) 26, 1115–40. Available at: http://www.escardio.org/NR/rdonlyres/8A2848B4-5DEB-41B9-9A0A-5B5A90494B64/0/ CHFFullTextehi205FVFW170505.pdf (accessed 14/08/07).
35. National Institute for Clinical Excellence. Chronic heart failure: management of chronic heart failure in adults in primary and secondary care (issued July 2003). Available at http://www.nice.org.uk/pdf/CG5NICEguideline.pdf (accessed 14/08/07).




1. Al-Khadra AS, Salem DN, Rand WM, Udelson JE, Smith JJ, Konstam MA. Antiplatelet agents and survival: a cohort analysis from the Studies of Left Ventricular Dysfunction (SOLVD) trial. J Am Coll Cardiol (1998) 31, 419-25.
2. Nguyen KN, Aursnes I, Kjekshus J. Interaction between enalapril and aspirin on mortality after acute myocardial infarction: subgroup analysis of the Cooperative New Scandanavian Enalapril Survival Study II (CONSENSUS II). Am J Cardiol (1997) 79, 115-19.
3. Peterson JG, Topol EJ, Sapp SK, Young JB, Lincoff AM, Lauer MS. Evaluation of the effects of aspirin combined with angiotensin-converting enzyme inhibitors in patients with coronary artery disease. Am J Med (2000) 109, 371-7.
4. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet (1993) 342, 821-8.
5. Leor J, Reicher-Reiss H, Goldbourt U, Boyko V, Gottlieb S, Battler A, Behar S. Aspirin and mortality in patients treated with angiotensin-converting enzyme inhibitors. A cohort study of 11,575 patients with coronary artery disease. J Am Coll Cardiol (1999) 33, 1920-5.
6. Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, Klein M, Lamas GA, Packer M, Rouleau J, Rouleau JL, Rutherford J, Wertheimer JH, Hawkins CM, on behalf of the SAVE Investigators. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med (1992) 327, 669-77.
7. The Heart Outcomes Prevention Evaluation Study Investigators. Effect of an angiotensin- converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med (2000) 342, 145-53.
8. Oosterga M, Anthonio RL, de Kam PJ, Kingma JH, Crijns HJ, van Gilst WH. Effects of aspirin on angiotensin-converting enzyme inhibition and left ventricular dilation one year after myocardial infarction. Am J Cardiol (1998) 81, 1178-81.
9. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet (1995) 345; 669-85.
10. Flather MD, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, Trop-Pedersen C, Ball S, Pogue J, Moyé L, Braunwald E, for the ACE-inhibitor Myocardial Infarction Collaborative.
Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet (2000) 335, 1575-81.
11. Latini R, Tognoni G, Maggioni AP, Baigent C, Braunwald E, Chen Z-M, Collins R, Flather M, Franzosi MG, Kjekshus J, Køber L, Liu L-S, Peto R, Pfeffer M, Pizzetti F, Santoro E, Sleight P, Swedberg K, Tavazzi L, Wang W, Yusuf S. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for myocardial infarction are similar in the presence and absence of aspirin. J Am Coll Cardiol (2000) 35 1801-7.
12. Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, Videbæk J, Cole DS, Auclert L, Pauly NC, Aliot E, Persson S, Camm AJ, for the Trandolapril Cardiac Evaluation (TRACE) Study Group. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med (1995) 333, 1670-6.
13. Ambrosioni E, Borghi C, Magnani B, for the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators. The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. N Engl J Med (1995) 332, 80-5.
14. Teo KK, Yusuf S, Pfeffer M, Kober L, Hall A, Pogue J, Latini R, Collins R, for the ACE Inhibitors Collaborative Group. Effects of long-term treatment with angiotensinconverting-enzyme inhibitors in the presence or absence of aspirin: a systematic review. Lancet (2002) 360, 1037-43.
2. Nguyen KN, Aursnes I, Kjekshus J. Interaction between enalapril and aspirin on mortality after acute myocardial infarction: subgroup analysis of the Cooperative New Scandanavian Enalapril Survival Study II (CONSENSUS II). Am J Cardiol (1997) 79, 115-19.
3. Peterson JG, Topol EJ, Sapp SK, Young JB, Lincoff AM, Lauer MS. Evaluation of the effects of aspirin combined with angiotensin-converting enzyme inhibitors in patients with coronary artery disease. Am J Med (2000) 109, 371-7.
4. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet (1993) 342, 821-8.
5. Leor J, Reicher-Reiss H, Goldbourt U, Boyko V, Gottlieb S, Battler A, Behar S. Aspirin and mortality in patients treated with angiotensin-converting enzyme inhibitors. A cohort study of 11,575 patients with coronary artery disease. J Am Coll Cardiol (1999) 33, 1920-5.
6. Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, Klein M, Lamas GA, Packer M, Rouleau J, Rouleau JL, Rutherford J, Wertheimer JH, Hawkins CM, on behalf of the SAVE Investigators. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med (1992) 327, 669-77.
7. The Heart Outcomes Prevention Evaluation Study Investigators. Effect of an angiotensin- converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med (2000) 342, 145-53.
8. Oosterga M, Anthonio RL, de Kam PJ, Kingma JH, Crijns HJ, van Gilst WH. Effects of aspirin on angiotensin-converting enzyme inhibition and left ventricular dilation one year after myocardial infarction. Am J Cardiol (1998) 81, 1178-81.
9. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet (1995) 345; 669-85.
10. Flather MD, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, Trop-Pedersen C, Ball S, Pogue J, Moyé L, Braunwald E, for the ACE-inhibitor Myocardial Infarction Collaborative.
Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet (2000) 335, 1575-81.
11. Latini R, Tognoni G, Maggioni AP, Baigent C, Braunwald E, Chen Z-M, Collins R, Flather M, Franzosi MG, Kjekshus J, Køber L, Liu L-S, Peto R, Pfeffer M, Pizzetti F, Santoro E, Sleight P, Swedberg K, Tavazzi L, Wang W, Yusuf S. Clinical effects of early angiotensin-converting enzyme inhibitor treatment for myocardial infarction are similar in the presence and absence of aspirin. J Am Coll Cardiol (2000) 35 1801-7.
12. Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, Videbæk J, Cole DS, Auclert L, Pauly NC, Aliot E, Persson S, Camm AJ, for the Trandolapril Cardiac Evaluation (TRACE) Study Group. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med (1995) 333, 1670-6.
13. Ambrosioni E, Borghi C, Magnani B, for the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators. The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. N Engl J Med (1995) 332, 80-5.
14. Teo KK, Yusuf S, Pfeffer M, Kober L, Hall A, Pogue J, Latini R, Collins R, for the ACE Inhibitors Collaborative Group. Effects of long-term treatment with angiotensinconverting-enzyme inhibitors in the presence or absence of aspirin: a systematic review. Lancet (2002) 360, 1037-43.
ACE inhibitors + Azathioprine
Anaemia has been seen in patients given azathioprine with enalapril or captopril. Leucopenia occasionally occurs when captopril is given with azathioprine.
Clinical evidence
(a) Anaemia
Nine out of 11 kidney transplant patients taking ACE inhibitors (enalapril or captopril) had a fall in their haematocrit from 34% to 27%, and a fall in their haemoglobin from 11.6 g/dL to 9.5 g/dL when ciclosporin was replaced by azathioprine. Two patients were switched back to ciclosporin, and had a prompt rise in their haematocrit. Another 10 patients taking both drugs similarly developed a degree of anaemia, when compared with 10 others not taking an ACE inhibitor (haematocrit of 33% compared with 41%, and a haemoglobin of 11.5 g/dL compared with 13.9 g/dL).1 A later study by the same group of workers (again in patients taking enalapril or captopril) confirmed these findings: however, no pharmacokinetic interaction was found between enalapril and azathioprine.2
(b) Leucopenia
A patient whose white cell count fell sharply when taking both captopril 50 mg daily and azathioprine 150 mg daily, did not develop leucopenia when each drug was given separately.3 Another patient who was given captopril (increased to 475 mg daily [sic] then reduced to 100 mg daily) immediately after discontinuing azathioprine, developed leucopenia. She was later successfully treated with captopril 4 to 6 mg daily [sic].4 Other patients have similarly shown leucopenia when given both drugs;5,6 in one case this did not recur when the patient was rechallenged with captopril
alone (at a lower dose).6
Mechanism
The anaemia appears to be due to suppression of erythropoietin by the ACE inhibitors, and azathioprine may cause patients to be more susceptible to this effect.2 The cause of the leucopenia is unknown. It may just be due to the additive effects of both drugs.
Importance and management
Anaemia caused by captopril and enalapril has been seen in kidney transplant patients and in dialysis patients (see ‘ACE inhibitors and Angiotensin II receptor antagonists + Epoetin’, p.25). The evidence that this effect can be potentiated by azathioprine is limited, but it would be prudent to monitor well if these drugs are used together.
The evidence that the concurrent use of ACE inhibitors and azathioprine increases the risk of leucopenia is also limited. However, the UK manufacturer of captopril recommends that captopril should be used with extreme caution in patients receiving immunosuppressants, especially if there is renal impairment. They advise that in such patients differential white blood cell counts should be performed before starting captopril, then every 2 weeks in the first 3 months of treatment, and periodically thereafter.
7 The UK manufacturers of a number of other ACE inhibitors also state in their prescribing information that the use of ACE inhibitors with cytostatic or immunosuppressive drugs may lead to an increased risk of leucopenia.
For other potential interactions with ACE inhibitors that might lead to an increased risk of leucopenia, see also ‘ACE inhibitors + Allopurinol’, p.13, and ‘ACE inhibitors + Procainamide’, p.33.
1. Gossmann J, Kachel H-G, Schoeppe W, Scheuermann E-H. Anemia in renal transplant recipients caused by concomitant therapy with azathioprine and angiotensin-converting enzyme inhibitors.
Transplantation (1993) 56, 585–9.
2. Gossmann J, Thürmann P, Bachmann T, Weller S, Kachel H-G, Schoeppe W, Scheuermann E-H. Mechanism of angiotensin converting enzyme inhibitor-related anemia in renal transplant recipients. Kidney Int (1996) 50, 973–8.
3. Kirchertz EJ, Gröne HJ, Rieger J, Hölscher M, Scheler F. Successful low dose captopril rechallenge following drug-induced leucopenia. Lancet (1981) i, 1363.
4. Case DB, Whitman HH, Laragh JH, Spiera H. Successful low dose captopril rechallenge following drug-induced leucopenia. Lancet (1981) i, 1362–3.
5. Elijovisch F, Krakoff LR. Captopril associated granulocytopenia in hypertension after renal transplantation. Lancet (1980), i, 927–8.
6. Edwards CRW, Drury P, Penketh A, Damluji SA. Successful reintroduction of captopril following neutropenia. Lancet (1981) i, 723.
7. Capoten (Captopril). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
7. Capoten (Captopril). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
ACE inhibitors + Beta blockers
The combination of an ACE inhibitor with a beta blocker is in established clinical use. Enhanced blood pressure-lowering effects occur, as would be expected. Although not all combinations have been studied, no clinically significant pharmacokinetic interactions appear to occur between the ACE inhibitors and beta blockers.
Clinical evidence
(a) Atenolol
In a double-blind, crossover study in hypertensive subjects, the combination of atenolol 50 mg once daily and enalapril 20 mg once daily increased the hypotensive effect of either drug alone, but the effect was 30 to 50% less than additive.1
(b) Bisoprolol
In a single-dose, placebo-controlled, crossover study in 16 healthy men, bisoprolol 5 mg given with imidapril 10 mg did not significantly influence the pharmacokinetics of its active metabolite imidaprilat, and the pharmacodynamic effects, including blood pressure and heart rate reductions, were mainly additive.2
(c) Propranolol
Propranolol 80 mg three times daily did not affect the pharmacokinetics of a single 20-mg dose of quinapril in 10 healthy subjects.3 The pharmacokinetics of ramipril 5 mg daily were unaffected by propranolol 40 mg twice daily.4 Similarly, the manufacturer of fosinopril reports that the bioavailability of fosinoprilat, its active metabolite, was not altered by propranolol.
5,6 Another study found no significant pharmacokinetic interaction between cilazapril 2.5 mg daily and propranolol 120 mg daily in healthy subjects, but the reductions in blood pressure were about doubled and long-lasting in healthy subjects and in patients with hypertension.7,8
Mechanism, importance and management
Both ACE inhibitors and beta blockers lower blood pressure by different mechanisms, and therefore the enhanced blood pressure-lowering effects of the combination would be expected. No pharmacokinetic interactions have been demonstrated. The combination of an ACE inhibitor and a beta blocker is clinically useful in a number of cardiovascular disorders.
1. Wing LMH, Chalmers JP, West MJ, Russell AE, Morris MJ, Cain MD, Bune AJC, Southgate DO. Enalapril and atenolol in essential hypertension: attenuation of hypotensive effects in combination. Clin Exp Hypertens (1988) 10, 119–33.
2. Breithaupt-Grögler K, Ungethüm W, Meurer-Witt B, Belz GG. Pharmacokinetic and dynamic interactions of the angiotensin-converting enzyme inhibitor imidapril with hydrochlorothiazide, bisoprolol and nilvadipine. Eur J Clin Pharmacol (2001) 57, 275–84.
3. Horvath AM, Pilon D, Caillé G, Colburn WA, Ferry JJ, Frank GJ, Lacasse Y, Olson SC. Multiple-dose propranolol administration does not influence the single dose pharmacokinetics of quinapril and its active metabolite (quinaprilat). Biopharm Drug Dispos (1990) 11, 191–6.
4. van Griensven JMT, Seibert-Grafe M, Schoemaker HC, Frölich M, Cohen AF. The pharmacokinetic and pharmacodynamic interactions of ramipril with propranolol. Eur J Clin Pharmacol (1993) 45, 255–60.
5. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
6. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
7. Belz GG, Essig J, Kleinbloesem CH, Hoogkamer JFW, Wiegand UW, Wellstein A. Interactions between cilazapril and propranolol in man; plasma drug concentrations, hormone and enzyme responses, haemodynamics, agonist dose-effect curves and baroreceptor reflex. Br J Clin Pharmacol (1988) 26, 547–56.
8. Belz GG, Essig J, Erb K, Breithaupt K, Hoogkamer JFW, Kneer J, Kleinbloesem CH. Pharmacokinetic and pharmacodynamic interactions between the ACE inhibitor cilazapril and β-adrenoceptor antagonist propranolol in healthy subjects and in hypertensive patients. Br J Clin Pharmacol (1989) 27, 317S–322S.
ACE inhibitors + Calcium-channel blockers
The combination of an ACE inhibitor and a dihydropyridine calcium-channel blocker is in established clinical use for hypertension, and, although only certain combinations have been studied, no clinically significant pharmacokinetic interactions appear to occur between the dihydropyridine-type calcium-channel blockers and ACE inhibitors
Clinical evidence
(a) Amlodipine
A study in 12 healthy subjects indicated that there was no pharmacokinetic interaction between single doses of amlodipine 5 mg and benazepril 10 mg.1
(b) Felodipine
No pharmacokinetic interaction occurred between single doses of felodipine 10 mg and ramipril 5 mg in healthy subjects. The blood pressure-lowering effect of the combination was greater, and ramipril attenuated the reflex tachycardia caused by felodipine.2
(c) Manidipine
In a single-dose crossover study in 18 healthy subjects, the concurrent use of manidipine 10 mg and delapril 30 mg did not significantly alter the pharmacokinetics of either drug or their main metabolites.3
(d) Nicardipine
In a study in 12 patients with hypertension taking enalapril 20 mg daily, the addition of nicardipine 30 mg three times daily for 2 weeks did not alter the pharmacokinetics of enalapril.4 The manufacturer of spirapril briefly noted in a review that the concurrent use of spirapril and nicardipine
increased spirapril plasma levels by about 25% and those of its active metabolite, spiraprilat, by about 45%. The bioavailability of nicardipine was reduced by 30%. It was assumed that the interaction took
place at the absorption site. However, the changes were not considered clinically relevant.5
(e) Nifedipine
No evidence of either a pharmacokinetic or adverse pharmacodynamic interaction was seen in 12 healthy subjects given single doses of nifedipine retard 20 mg and lisinopril 20 mg; the effects on blood pressure were additive. 6 Similarly, there was no pharmacokinetic interaction between single doses of slow-release nifedipine 20 mg and benazepril 10 mg in healthy subjects; the effects on blood pressure were additive and the tachycardic effect of nifedipine was attenuated by benazepril.7 The manufacturer of fosinopril notes that the bioavailability of fosinoprilat, the active metabolite, was not altered by nifedipine.8,9 Similarly, the manufacturer of moexipril notes that no clinically important pharmacokinetic interaction occurred with nifedipine in healthy subjects.10
(f) Nilvadipine
In a single-dose, placebo-controlled, crossover study in 16 healthy subjects, no pharmacokinetic interaction occurred between nilvadipine 8 mg and imidapril 10 mg, and the pharmacodynamic effects, including the reduction in blood pressure and the decrease in total peripheral resistance,
were mostly additive.11
Mechanism
No pharmacokinetic interactions are expected. Enhanced blood pressurelowering effects occur, as would be expected.
Importance and management
No important pharmacokinetic interactions have been demonstrated. The combination of an ACE inhibitor and a dihydropyridine calcium-channel blocker is clinically useful in the treatment of hypertension. A number of products combining an ACE inhibitor with a calcium-channel blocker are available. It is generally advised that these combination products are only used in patients who have already been stabilised on the individual components in the same proportions.
1. Sun JX, Cipriano A, Chan K, John VA. Pharmacokinetic interaction study between benazepril and amlodipine in healthy subjects. Eur J Clin Pharmacol (1994) 47, 285–9.
2. Bainbridge AD, MacFadyen RJ, Lees KR, Reid JL. A study of the acute pharmacodynamic interaction of ramipril and felodipine in normotensive subjects. Br J Clin Pharmacol (1991) 31, 148–53.
3. Stockis A, Gengler C, Goethals F, Jeanbaptiste B, Lens S, Poli G, Acerbi D. Single oral dose pharmacokinetic interaction study of manidipine and delapril in healthy volunteers. Arzneimittelforschung (2003) 53, 627–34.
4. Donnelly R, Elliott HL, Reid JL. Nicardipine combined with enalapril in patients with essential hypertension. Br J Clin Pharmacol (1986) 22, 283S–287S.
5. Grass P, Gerbeau C, Kutz K. Spirapril: pharmacokinetic properties and drug interactions. Blood Pressure (1994) 3 (Suppl 2), 7–13.
6. Lees KR, Reid JL. Lisinopril and nifedipine: no acute interaction in normotensives. Br J Clin Pharmacol (1988) 25, 307–13.
7. Jakobsen J, Glaus L, Graf P, Degen P, Maurice NP, Bellet M, Ménard J. Unmasking of the hypotensive effect of nifedipine in normotensives by addition of the angiotensin converting enzyme inhibitor benazepril. J Hypertens (1992) 10, 1045–51.
8. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
9. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
10. Perdix (Moexipril hydrochloride). Schwarz Pharma Ltd. UK Summary of product characteristics, July 2006.
11. Breithaupt-Grögler K, Ungethüm W, Meurer-Witt B, Belz GG. Pharmacokinetic and dynamic interactions of the angiotensin-converting enzyme inhibitor imidapril with hydrochlorothiazide, bisoprolol and nilvadipine. Eur J Clin Pharmacol (2001) 57, 275–84.
8. Staril (Fosinopril sodium). E. R. Squibb & Sons Ltd. UK Summary of product characteristics, June 2005.
9. Monopril (Fosinopril sodium). Bristol-Myers Squibb Company. US Prescribing information, July 2003.
10. Perdix (Moexipril hydrochloride). Schwarz Pharma Ltd. UK Summary of product characteristics, July 2006.
11. Breithaupt-Grögler K, Ungethüm W, Meurer-Witt B, Belz GG. Pharmacokinetic and dynamic interactions of the angiotensin-converting enzyme inhibitor imidapril with hydrochlorothiazide, bisoprolol and nilvadipine. Eur J Clin Pharmacol (2001) 57, 275–84.
ACE inhibitors + Capsaicin
An isolated report describes a woman taking an ACE inhibitor who developed a cough each time she used a topical cream containing capsaicin.
Clinical evidence, mechanism, importance and management
A 53-year-old woman who had been taking an unnamed ACE inhibitor for several years, complained of cough each time she applied Axsain, a cream containing capsaicin 0.075%, to her lower extremities. Whether this reaction would have occurred without the ACE inhibitor was not determined,1 but cough is a recognised adverse effect of ACE inhibitors and pre-treatment with an ACE inhibitor has been shown to enhance the cough caused by inhaled capsaicin.1 This potential interaction is probably of little general clinical importance.
1. Hakas JF. Topical capsaicin induces cough in patient receiving ACE inhibitor. Ann Allergy (1990) 65, 322.
ACE inhibitors + Clonidine
Potentiation of the antihypertensive effect of clonidine by ACE inhibitors can be clinically useful.1 However, limited evidence suggests that the effects of captopril may be delayed when patients are switched from clonidine.2 Note that sudden withdrawal of clonidine may cause rebound hypertension.
1. Catapres Tablets (Clonidine hydrochloride). Boehringer Ingelheim Ltd. UK Summary of product characteristics, May 2006.
2. Gröne H-J, Kirchertz EJ, Rieger J. Mögliche Komplikationen und Probleme der Captopriltherapie bei Hypertonikern mit ausgeprägten Gefäßschäden. Therapiewoche (1981) 31, 5280–7.
1. Catapres Tablets (Clonidine hydrochloride). Boehringer Ingelheim Ltd. UK Summary of product characteristics, May 2006.
2. Gröne H-J, Kirchertz EJ, Rieger J. Mögliche Komplikationen und Probleme der Captopriltherapie bei Hypertonikern mit ausgeprägten Gefäßschäden. Therapiewoche (1981) 31, 5280–7.
ACE inhibitors + Colloids
Acute hypotension has been seen in a few patients taking enalapril when they were given a rapid infusion of albumin-containing stable plasma protein solution (SPPS). Another case occurred in an infant taking captopril when given albumin 4%. A few other cases have been described with gelatin-type colloids in patients taking ACE inhibitors (cilazapril, enalapril, lisinopril).
Clinical evidence
(a) Albumin
A woman taking enalapril 10 mg in the morning, underwent surgery for groin lymph node resection under spinal and general anaesthesia. When she was given a rapid infusion of 500 mL of the albumin solution, stable plasma protein solution (SPPS, Commonwealth Serum Laboratories, Melbourne, Australia), her pulse rose to 90 to 100 bpm and systolic blood pressure fell from 100 to 60 mmHg and a red flush was noted on all exposed skin. The blood pressure was controlled at 90 to 95 mmHg with metaraminol 4.5 mg, given over 10 minutes. When the SPPS was finished, the blood pressure and pulse rate spontaneously restabilised.1 SPPS is a 5% plasma protein solution prepared by the cold ethanol fractionation process and pasteurisation from human plasma (volunteer donors). It contains sodium octanoate as a stabiliser.1 Two very similar cases have been recorded in patients taking enalapril when given SPPS.2,3 The manufacturer of SPPS notes that captopril has also been involved in this hypotensive interaction A 20-month-old infant taking captopril was haemodynamically stable for 35 minutes after induction of anaesthesia while awaiting a donor kidney, but then developed hypotension after a bolus dose of 20 mL of albumin 4% (Albumex) was given. This was reversed with dopamine infusion.5
(b) Gelatin-based colloids
A report describes 3 cases of severe hypotension in patients taking ACE inhibitors (lisinopril, enalapril) while undergoing joint replacement surgery, and after they had been given a gelatin-based plasma expander (Gelofusin), which contains 4% succinylated gelatin in saline. The hypotension was resistant to ephedrine and methoxamine, and responded to adrenaline or dobutamine, which was required for 24 hours and 3 days in two cases. Anaphylactoid reactions were excluded as a cause of the hypotension.6 In another similar case, a patient taking cilazapril developed hypotension refractory to sodium chloride 0.9% after induction of anaesthesia, and this worsened when a gelatin-type colloid (Gelafundina) was given.7
Mechanism
Not fully established, but it is believed that SPPS contains low levels of pre-kallikrein activator, which stimulates the production of bradykinin, which can cause vasodilatation and hypotension. Normally the bradykinin is destroyed by kininase II (ACE), but this is delayed by the ACE inhibitor so that the hypotensive effects are exaggerated and prolonged.3,8 In the case with albumin 4%, a sample of the albumin used was analysed, and it was found to contain less prekallikrein activating factor than maximum permissible levels.5 It was suggested that the infusion of gelatin-based colloids
somehow resulted in raised plasma kinin levels associated with inhibition of ACE.6
Not fully established, but it is believed that SPPS contains low levels of pre-kallikrein activator, which stimulates the production of bradykinin, which can cause vasodilatation and hypotension. Normally the bradykinin is destroyed by kininase II (ACE), but this is delayed by the ACE inhibitor so that the hypotensive effects are exaggerated and prolonged.3,8 In the case with albumin 4%, a sample of the albumin used was analysed, and it was found to contain less prekallikrein activating factor than maximum permissible levels.5 It was suggested that the infusion of gelatin-based colloids
somehow resulted in raised plasma kinin levels associated with inhibition of ACE.6
Importance and management
The interaction with SPPS would appear to be established and of clinical importance, and would apply to all ACE inhibitors. The author of one report suggested that if rapid expansion of intravascular volume is needed in patients taking ACE inhibitors, an artificial colloid might be a safer choice than SPPS.1 The manufacturer of SPPS also recommended using an alternative plasma volume expander, including other albumin solutions.4 It should be noted that following these reports SPPS was withdrawn from the Australasian market.9 However, note that a case has also occurred with albumin 4%, and cases have also been attributed to synthetic colloid solutions containing gelatin. It may be that this is just an unpredictable effect of colloids in patients taking ACE inhibitors. See also ‘Anaesthetics, general + Antihypertensives’, p.94, for discussion of the marked hypotension sometimes seen during induction of anaesthesia in patients taking ACE inhibitors.
1. McKenzie AJ. Possible interaction between SPPS and enalapril. Anaesth Intensive Care (1990) 18, 124–6.
2. Young K. Enalapril and SPPS. Anaesth Intensive Care (1990) 18, 583.
3. Young K. Hypotension from the interaction of ACE inhibitors with stable plasma protein solution. Anaesthesia (1993) 48, 356.
4. Schiff P. SPPS, hypotension and ACE inhibitors. Med J Aust (1992) 156, 363.
5. Fong SY, Hansen TG. Perioperative hypotension following plasma volume expansion with albumin in an angiotensin-converting enzyme inhibited infant. Br J Anaesth (2000) 84, 537–8.
6. Powell CG, Unsworth DJ, McVey FK. Severe hypotension associated with angiotensin-converting enzyme inhibition in anaesthesia. Anaesth Intensive Care (1998) 26, 107–9.
7. Barber L, Barrio J, de Rojas MD, Ibañez F, Añó C, Alepuz R, Montero R. Hipotensión refractaria y sostenida durante una anestesia general asociada al tratamiento crónico con inhibidores de la enzima conversiva de la angiotensina. Rev Esp Anestesiol Reanim. (2001) 48, 34–7.
8. Bönner G, Preis S, Schunk U, Toussaint C, Kaufmann W. Hemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans. J Cardiovasc Pharmacol (1990) 15 (Suppl 6), S46–S56.
9. McKenzie AJ. ACE inhibitors, colloid infusions and anaesthesia. Anaesth Intensive Care (1998) 26, 330.



